

Morphometry and network-based atrophy patterns in SCN1A-related Dravet syndrome
- PMID: 37344172
- PMCID: PMC10431750
- DOI: 10.1093/cercor/bhad224
Morphometry and network-based atrophy patterns in SCN1A-related Dravet syndrome
Abstract
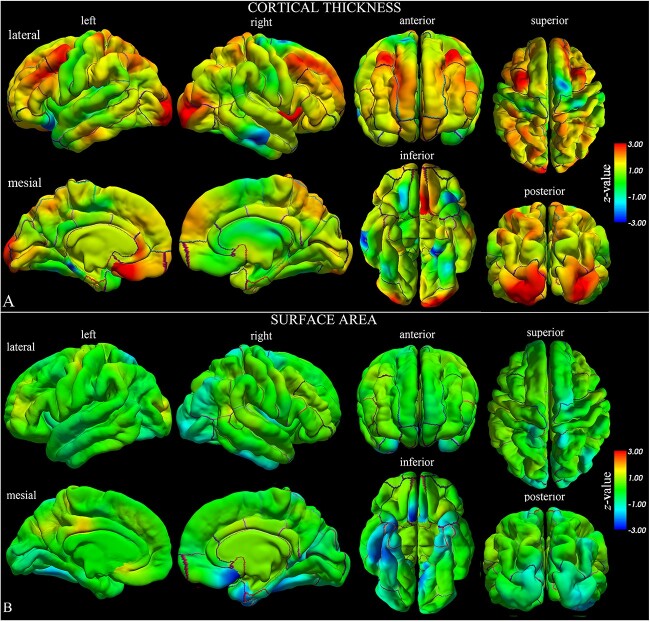
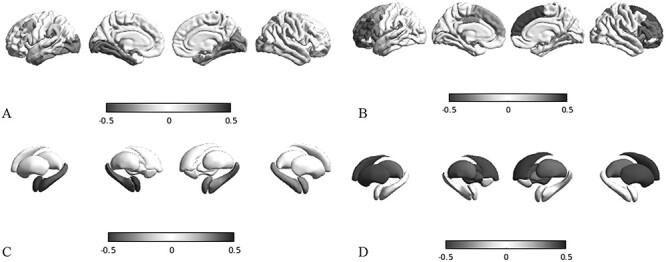
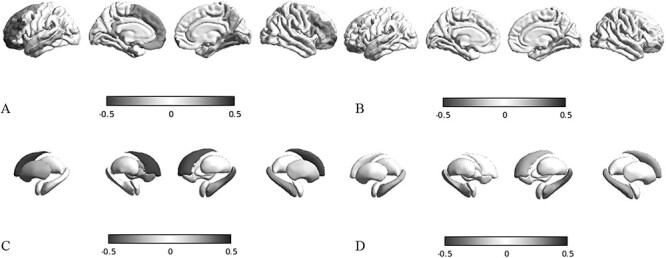
Mutations of the voltage-gated sodium channel SCN1A gene (MIM#182389) are among the most clinically relevant epilepsy-related genetic mutations and present variable phenotypes, from the milder genetic epilepsy with febrile seizures plus to Dravet syndrome, a severe developmental and epileptic encephalopathy. Qualitative neuroimaging studies have identified malformations of cortical development in some patients and mild atrophic changes, partially confirmed by quantitative studies. Precise correlations between MRI findings and clinical variables have not been addressed. We used morphometric methods and network-based models to detect abnormal brain structural patterns in 34 patients with SCN1A-related epilepsy, including 22 with Dravet syndrome. By measuring the morphometric characteristics of the cortical mantle and volume of subcortical structures, we found bilateral atrophic changes in the hippocampus, amygdala, and the temporo-limbic cortex (P-value < 0.05). By correlating atrophic patterns with brain connectivity profiles, we found the region of the hippocampal formation as the epicenter of the structural changes. We also observed that Dravet syndrome was associated with more severe atrophy patterns with respect to the genetic epilepsy with febrile seizures plus phenotype (r = -0.0613, P-value = 0.03), thus suggesting that both the underlying mutation and seizure severity contribute to determine atrophic changes.
Keywords: Dravet syndrome; SCN1A mutation; epilepsy; hippocampus; limbic formation.
© The Author(s) 2023. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
Similar articles
-
SCN1A-deficient excitatory neuronal networks display mutation-specific phenotypes.Brain. 2023 Dec 1;146(12):5153-5167. doi: 10.1093/brain/awad245. Brain. 2023. PMID: 37467479 Free PMC article.
-
Outcomes and comorbidities of SCN1A-related seizure disorders.Epilepsy Behav. 2019 Jan;90:252-259. doi: 10.1016/j.yebeh.2018.09.041. Epub 2018 Dec 5. Epilepsy Behav. 2019. PMID: 30527252
-
Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies.Epilepsia. 2019 Dec;60 Suppl 3:S2-S7. doi: 10.1111/epi.16054. Epilepsia. 2019. PMID: 31904125 Review.
-
Fine Mapping of a Dravet Syndrome Modifier Locus on Mouse Chromosome 5 and Candidate Gene Analysis by RNA-Seq.PLoS Genet. 2016 Oct 21;12(10):e1006398. doi: 10.1371/journal.pgen.1006398. eCollection 2016 Oct. PLoS Genet. 2016. PMID: 27768696 Free PMC article.
-
Dravet syndrome and Dravet syndrome-like phenotype: a systematic review of the SCN1A and PCDH19 variants.Neurogenetics. 2021 May;22(2):105-115. doi: 10.1007/s10048-021-00644-7. Epub 2021 May 3. Neurogenetics. 2021. PMID: 33937968
Cited by
-
Morphometric network-based abnormalities correlate with psychiatric comorbidities and gene expression in PCDH19-related developmental and epileptic encephalopathy.Transl Psychiatry. 2024 Jan 18;14(1):35. doi: 10.1038/s41398-024-02753-x. Transl Psychiatry. 2024. PMID: 38238304 Free PMC article.
References
-
- Baasch AL, Hüning I, Gilissen C, Klepper J, Veltman JA, Gillessen-Kaesbach G, Hoischen A, Lohmann K. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia. 2014:55(4):25–29. - PubMed
-
- Barba C, Parrini E, Coras R, Galuppi A, Craiu D, Kluger G, Parmeggiani A, Pieper T, Schmitt-Mechelke T, Striano P, et al. Co-occurring malformations of cortical development and SCN1A gene mutations. Epilepsia. 2014:55(7):1009–1019. - PubMed
