

Single-cell quantification of ribosome occupancy in early mouse development
- PMID: 37344592
- PMCID: PMC10307641
- DOI: 10.1038/s41586-023-06228-9
Single-cell quantification of ribosome occupancy in early mouse development
Abstract
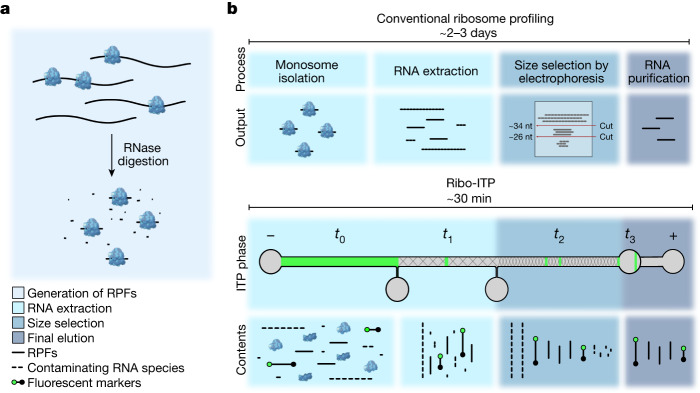
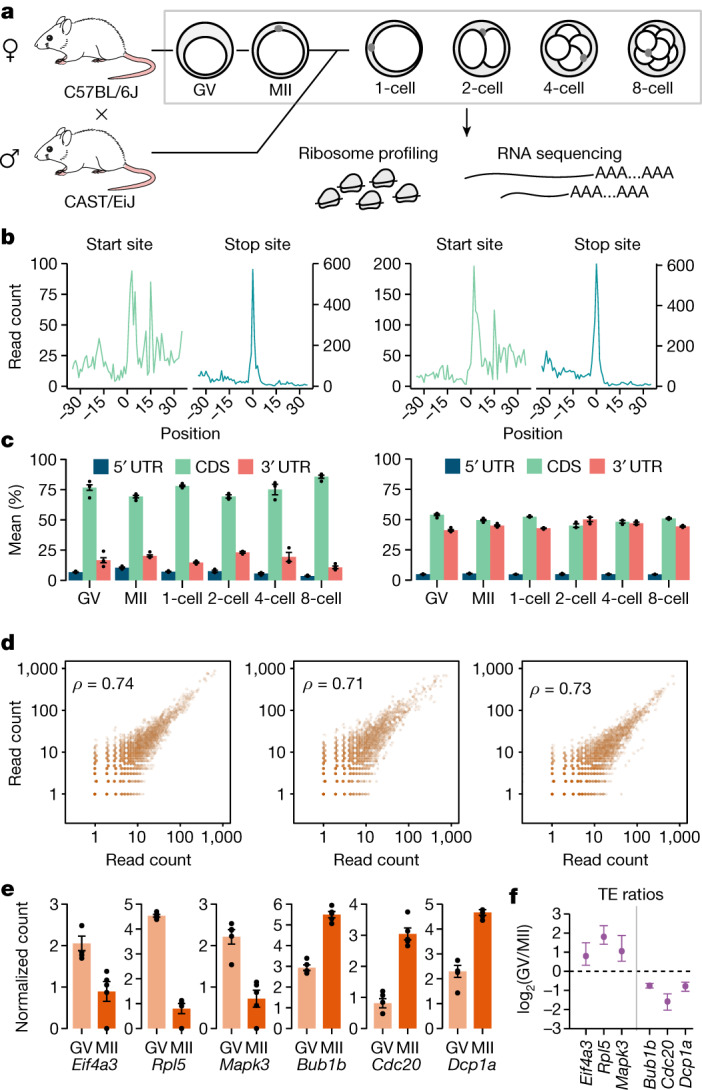
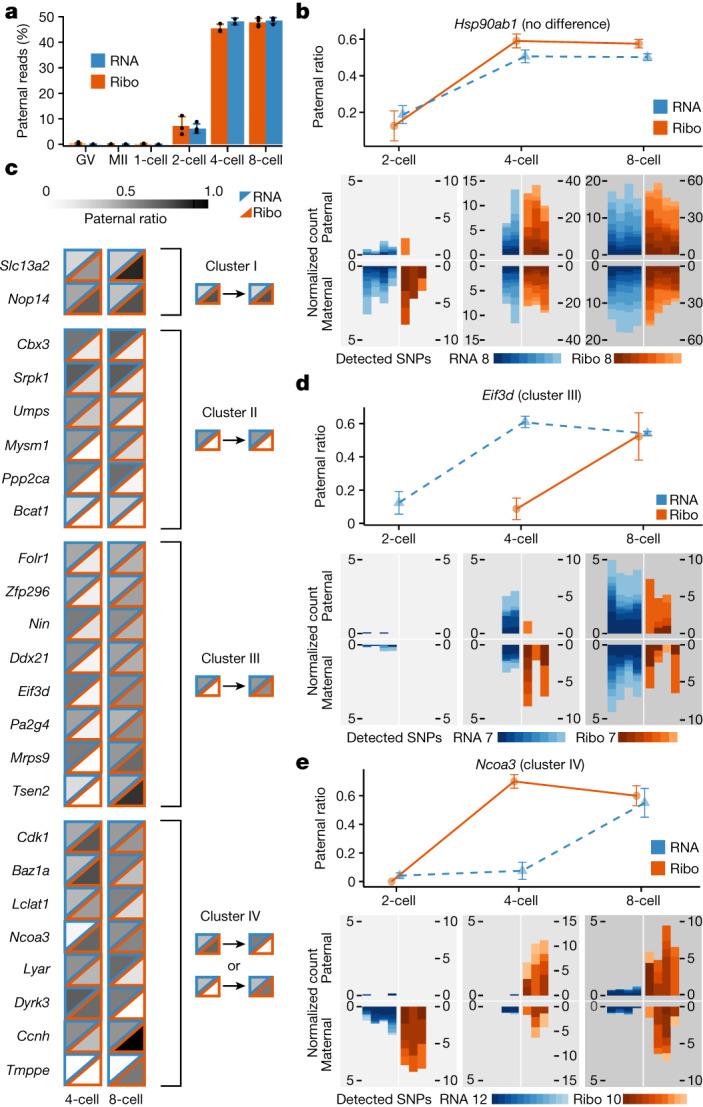
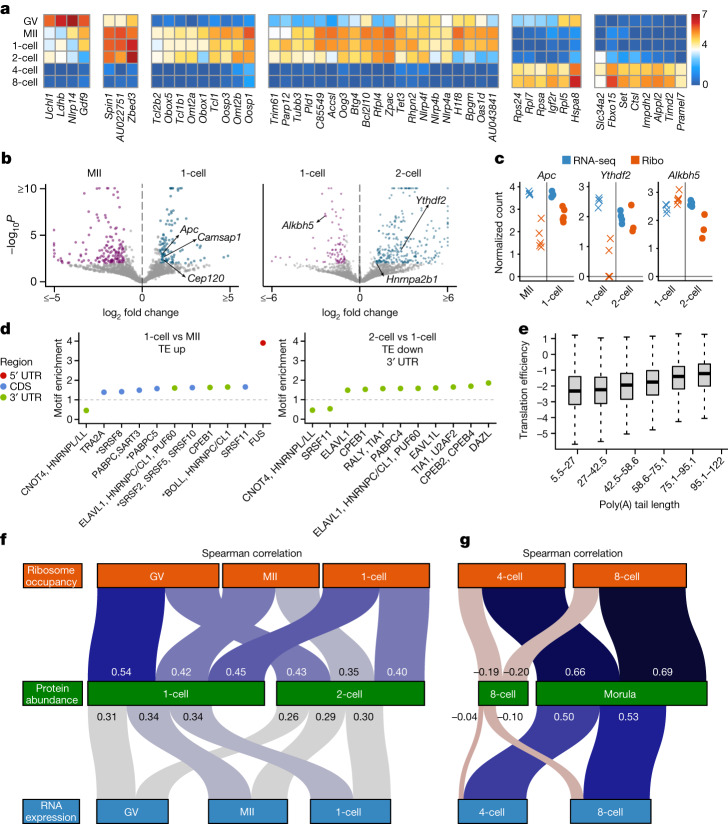
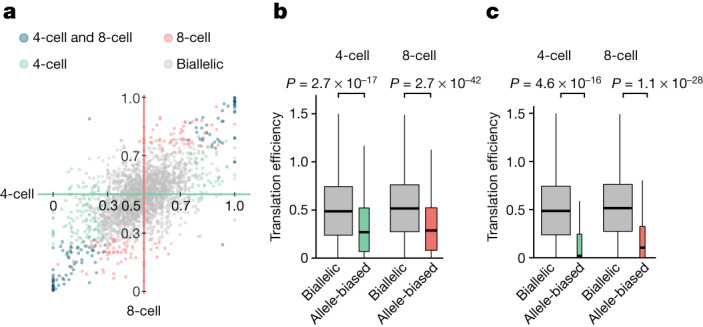
Translation regulation is critical for early mammalian embryonic development1. However, previous studies had been restricted to bulk measurements2, precluding precise determination of translation regulation including allele-specific analyses. Here, to address this challenge, we developed a novel microfluidic isotachophoresis (ITP) approach, named RIBOsome profiling via ITP (Ribo-ITP), and characterized translation in single oocytes and embryos during early mouse development. We identified differential translation efficiency as a key mechanism regulating genes involved in centrosome organization and N6-methyladenosine modification of RNAs. Our high-coverage measurements enabled, to our knowledge, the first analysis of allele-specific ribosome engagement in early development. These led to the discovery of stage-specific differential engagement of zygotic RNAs with ribosomes and reduced translation efficiency of transcripts exhibiting allele-biased expression. By integrating our measurements with proteomics data, we discovered that ribosome occupancy in germinal vesicle-stage oocytes is the predominant determinant of protein abundance in the zygote. The Ribo-ITP approach will enable numerous applications by providing high-coverage and high-resolution ribosome occupancy measurements from ultra-low input samples including single cells.
© 2023. The Author(s).
Conflict of interest statement
US patent application no. 63/286,531, entitled ‘Ribosome profiling via isotachophoresis’ by C.C. and C.M.H., and international patent application no. PCT/US2022/080982 based on US serial no. 63/286,531, entitled ‘Ribosome profiling via isotachophoresis’ by C.C. and C.M.H. were filed in the Name of Board of Regents, The University of Texas System et al.
Figures











References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
