

The computational capabilities of many-to-many protein interaction networks
- PMID: 37348461
- PMCID: PMC10318606
- DOI: 10.1016/j.cels.2023.05.001
The computational capabilities of many-to-many protein interaction networks
Abstract
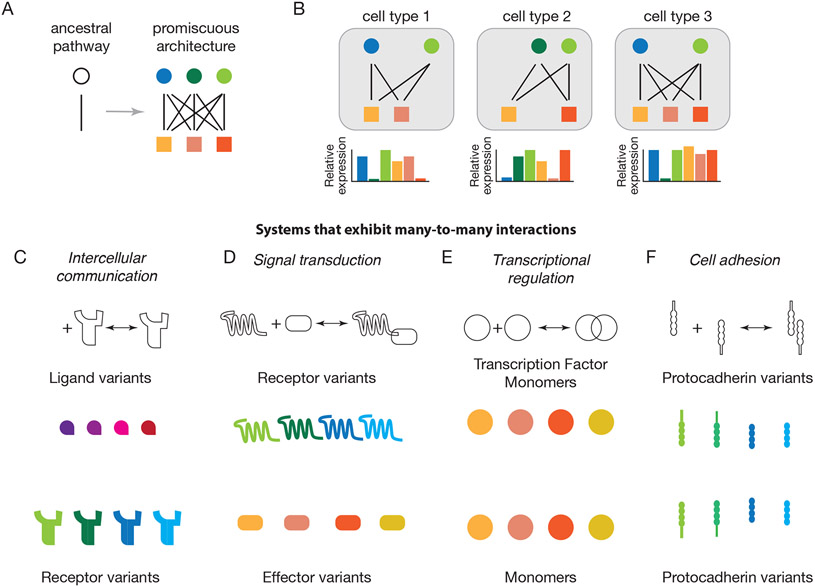
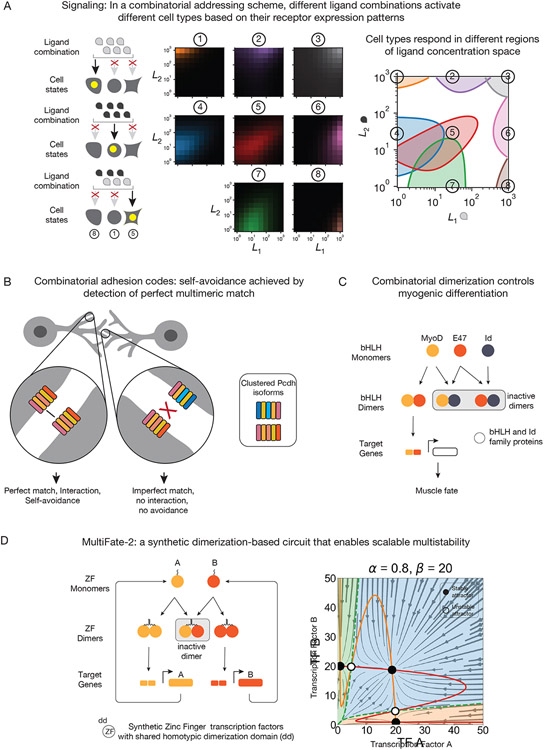
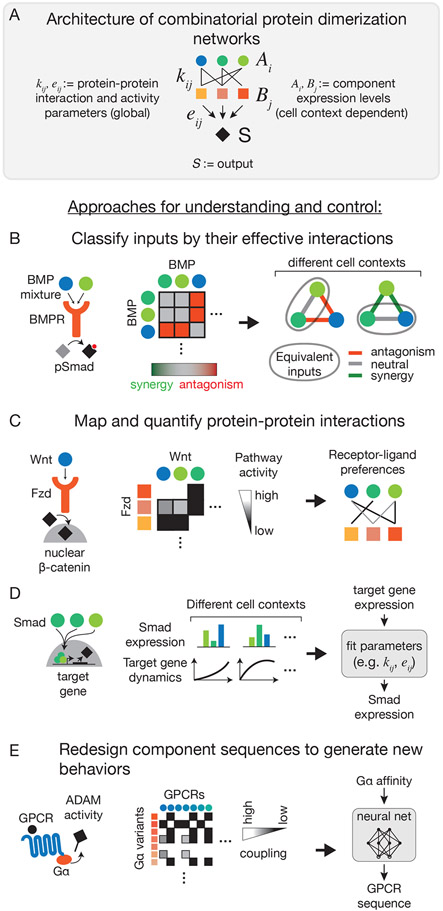
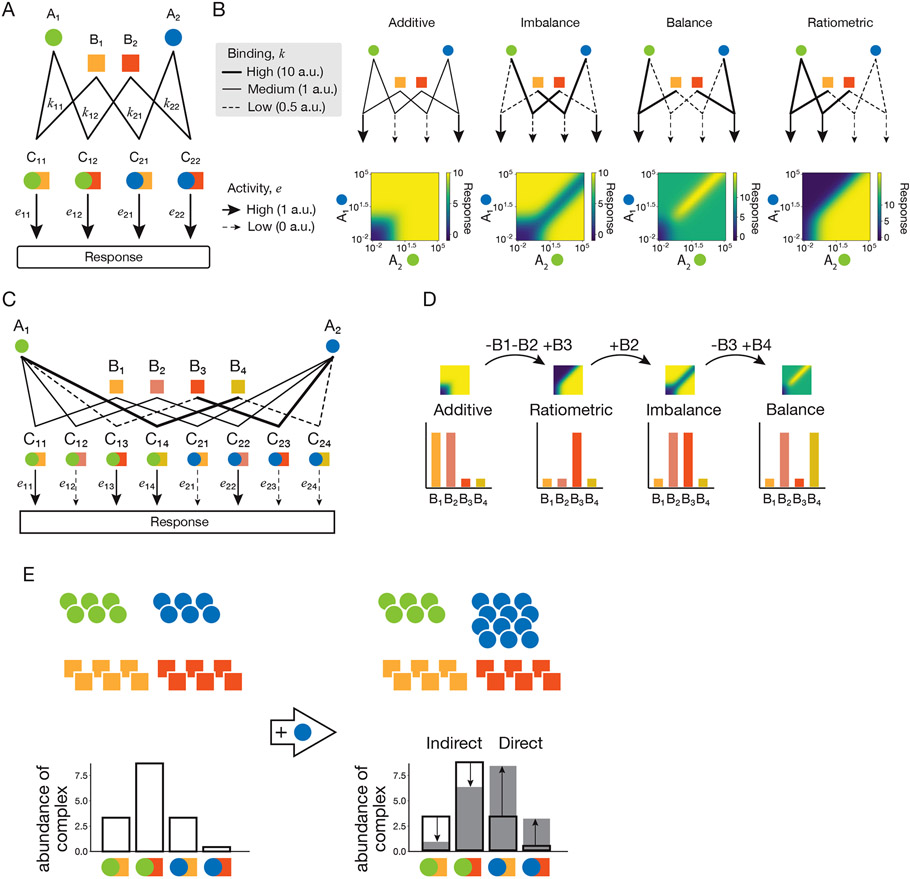
Many biological circuits comprise sets of protein variants that interact with one another in a many-to-many, or promiscuous, fashion. These architectures can provide powerful computational capabilities that are especially critical in multicellular organisms. Understanding the principles of biochemical computations in these circuits could allow more precise control of cellular behaviors. However, these systems are inherently difficult to analyze, due to their large number of interacting molecular components, partial redundancies, and cell context dependence. Here, we discuss recent experimental and theoretical advances that are beginning to reveal how promiscuous circuits compute, what roles those computations play in natural biological contexts, and how promiscuous architectures can be applied for the design of synthetic multicellular behaviors.
Keywords: biological circuit; biological computation; combinatorial protein interactions; molecular computation; promiscuous protein interactions; protein networks; signal transduction.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.B.E. is a scientific advisory board member or consultant at TeraCyte, Primordium, and Spatial Genomics. Y.E.A. is a scientific advisory board member or consultant at TeraCyte.
Figures
References
-
- Carretero-Paulet L, Galstyan A, Roig-Villanova I, Martínez-García JF, Bilbao-Castro JR, and Robertson DL (2010). Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 153, 1398–1412. 10.1104/pp.110.153593. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
