

Positional influence on cellular transcriptional identity revealed through spatially segmented single-cell transcriptomics
- PMID: 37348462
- PMCID: PMC10424188
- DOI: 10.1016/j.cels.2023.05.003
Positional influence on cellular transcriptional identity revealed through spatially segmented single-cell transcriptomics
Abstract
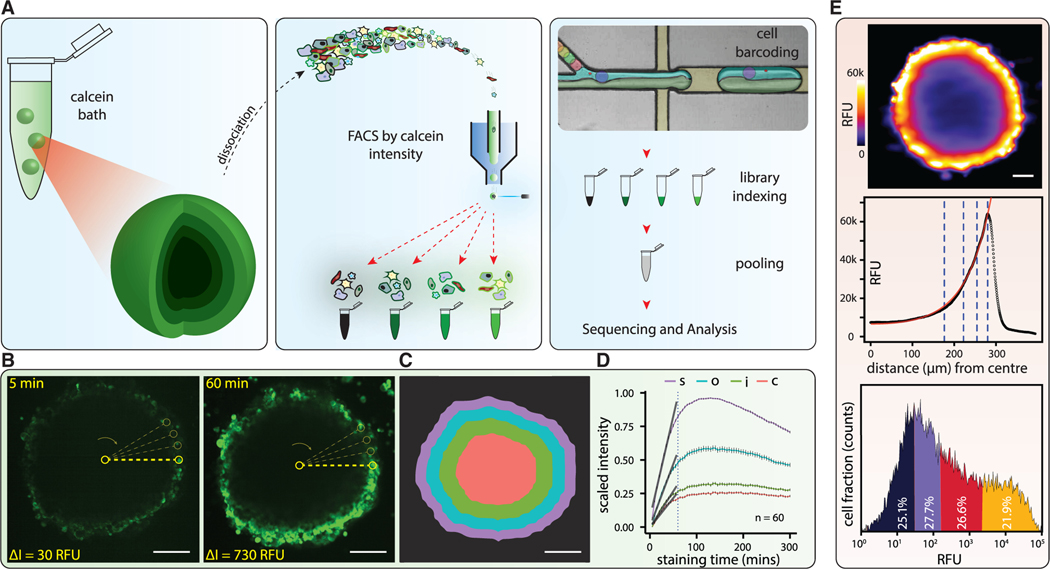
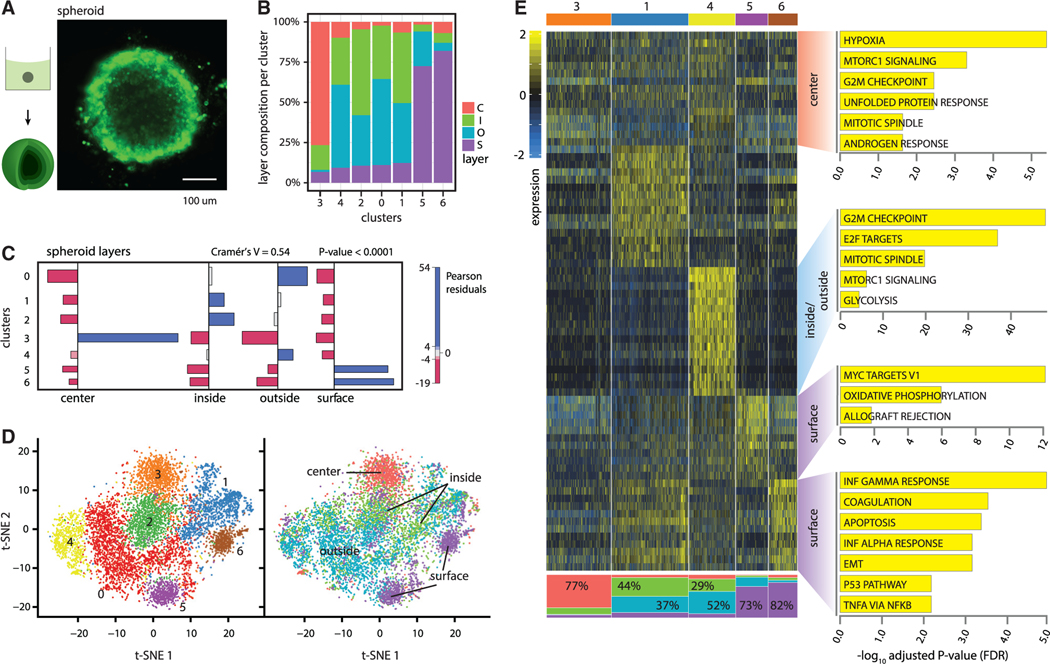
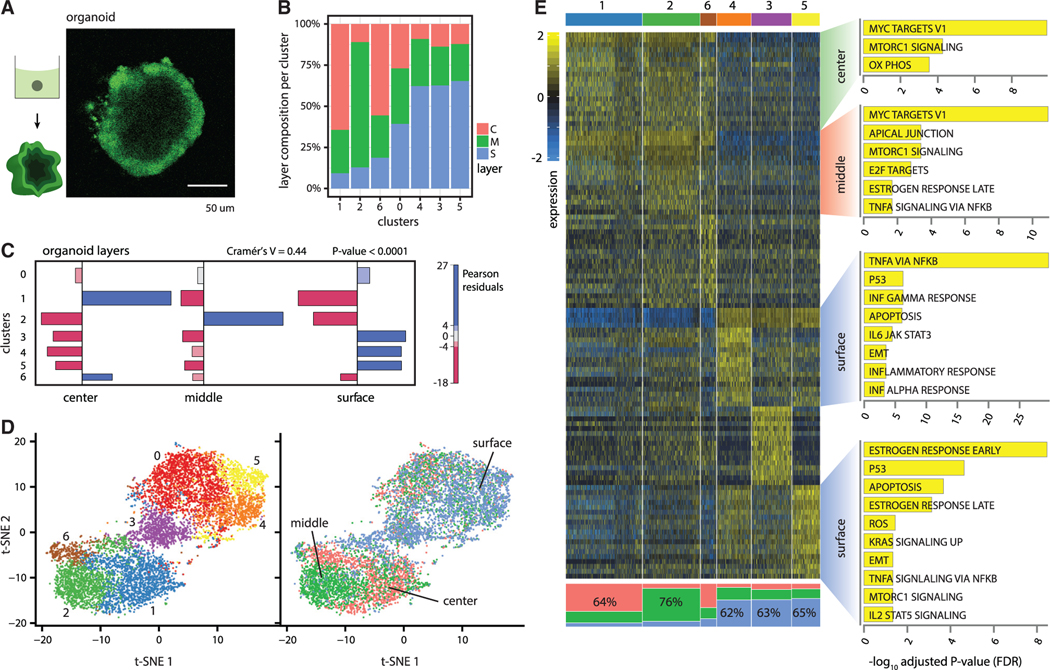
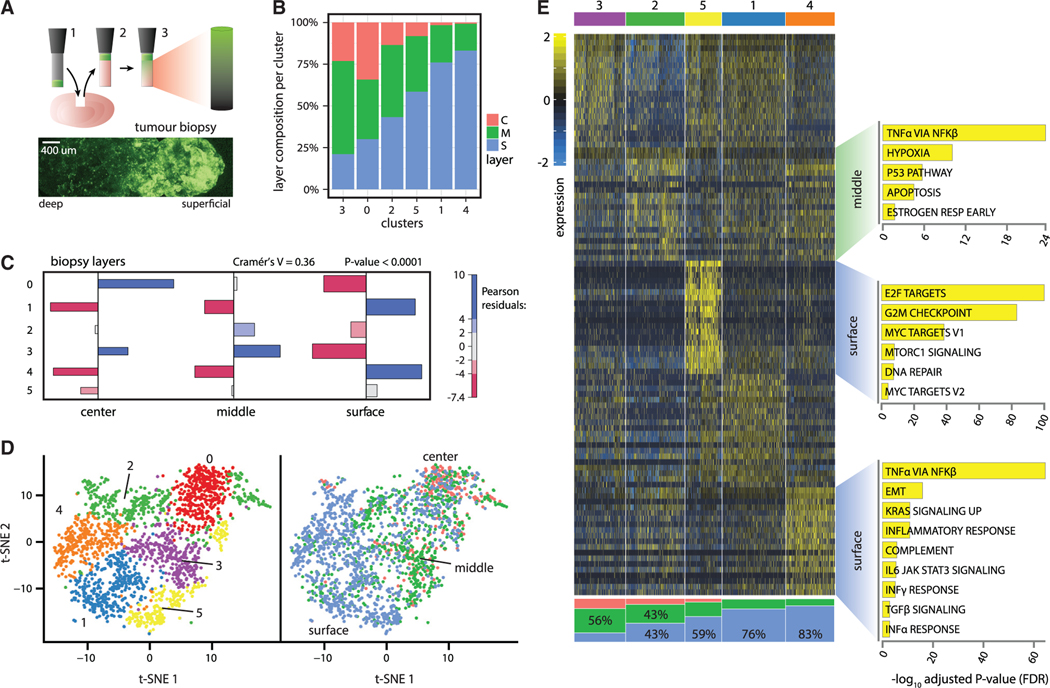
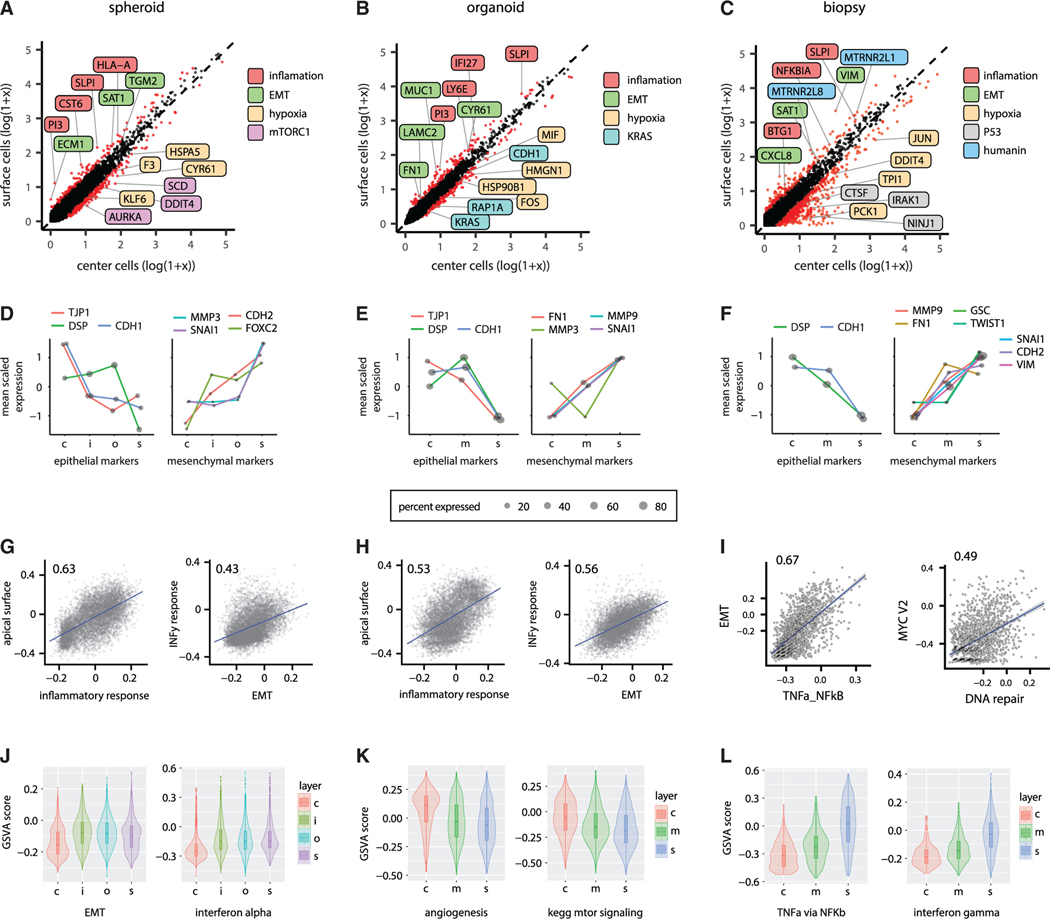
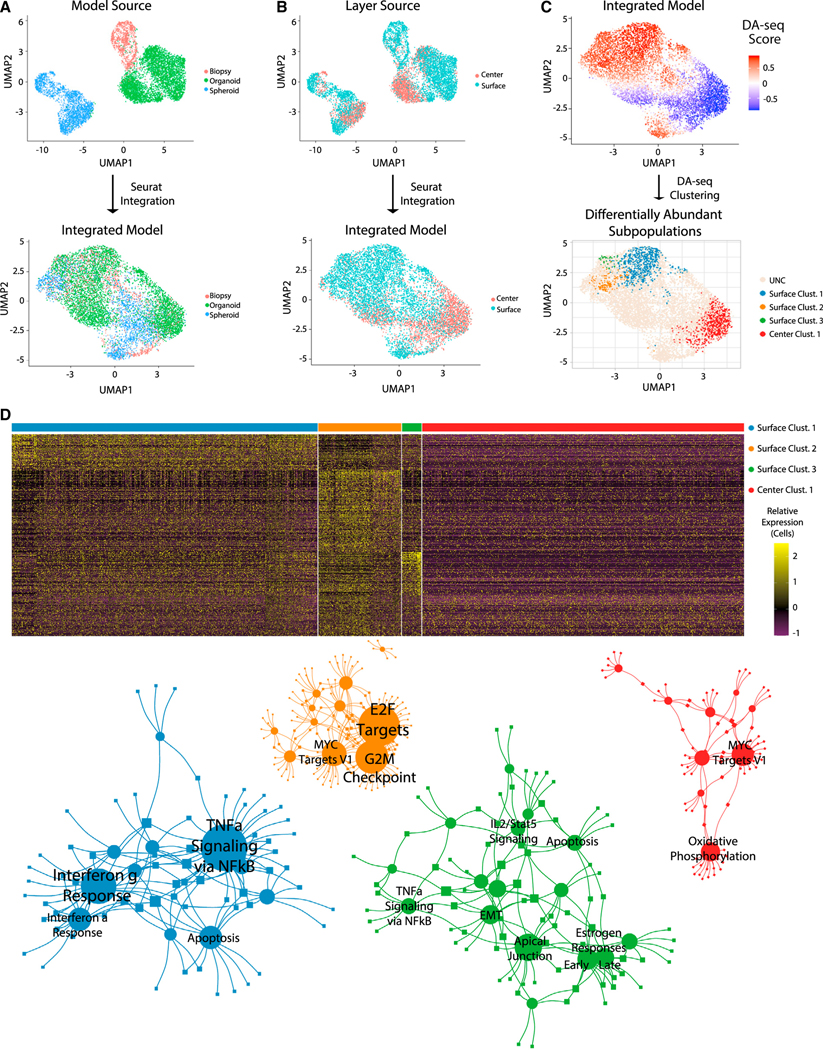
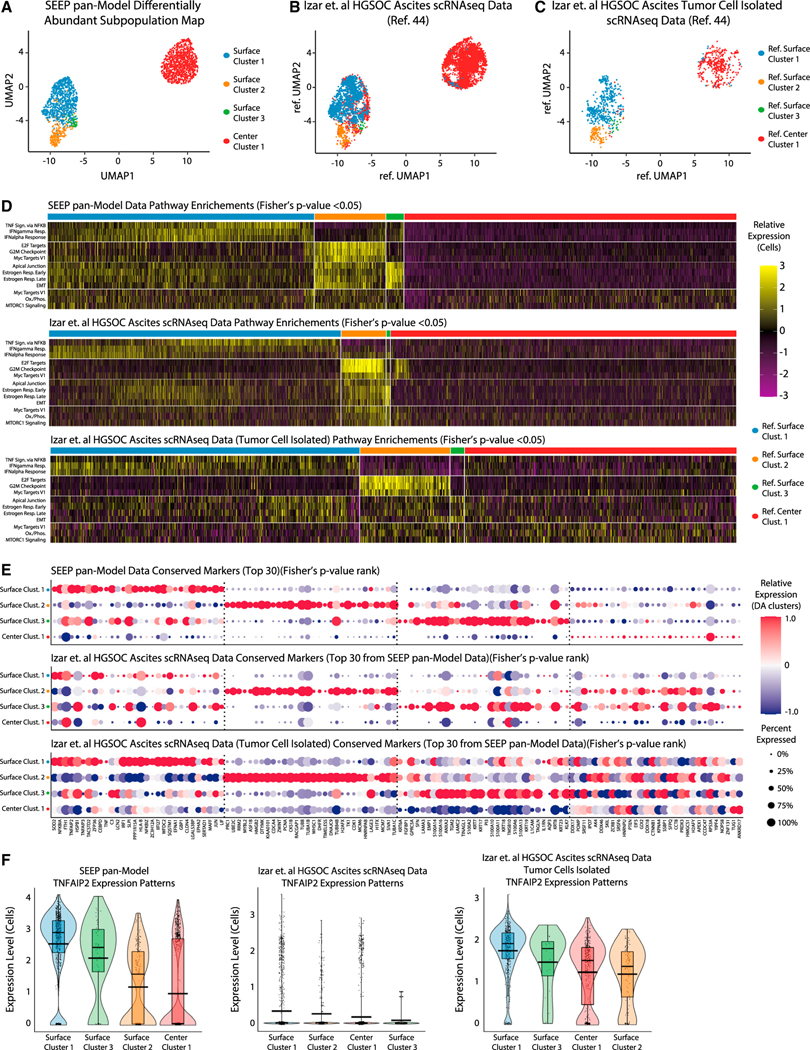
Single-cell RNA sequencing (scRNA-seq) is a powerful technique for describing cell states. Identifying the spatial arrangement of these states in tissues remains challenging, with the existing methods requiring niche methodologies and expertise. Here, we describe segmentation by exogenous perfusion (SEEP), a rapid and integrated method to link surface proximity and environment accessibility to transcriptional identity within three-dimensional (3D) disease models. The method utilizes the steady-state diffusion kinetics of a fluorescent dye to establish a gradient along the radial axis of disease models. Classification of sample layers based on dye accessibility enables dissociated and sorted cells to be characterized by transcriptomic and regional identities. Using SEEP, we analyze spheroid, organoid, and in vivo tumor models of high-grade serous ovarian cancer (HGSOC). The results validate long-standing beliefs about the relationship between cell state and position while revealing new concepts regarding how spatially unique microenvironments influence the identity of individual cells within tumors.
Keywords: RNA sequencing; genomics; oncology; ovarian cancer; scRNA-seq; single-cell transcriptomics; spatial transcriptomics; spatially resolved transcriptomics.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Bissell MJ, Kenny PA, and Radisky DC (2005). Microenvironmentalregulators of tissue structure and function also regulate tumor induction and progression: the role of extracellular matrix and its degrading enzymes. Cold Spring Harb. Symp. Quant. Biol. 70, 343–356. 10.1101/sqb.2005.70.013. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
