

CSF proteomics in autosomal dominant Alzheimer's disease highlights parallels with sporadic disease
- PMID: 37348871
- PMCID: PMC10629764
- DOI: 10.1093/brain/awad213
CSF proteomics in autosomal dominant Alzheimer's disease highlights parallels with sporadic disease
Abstract
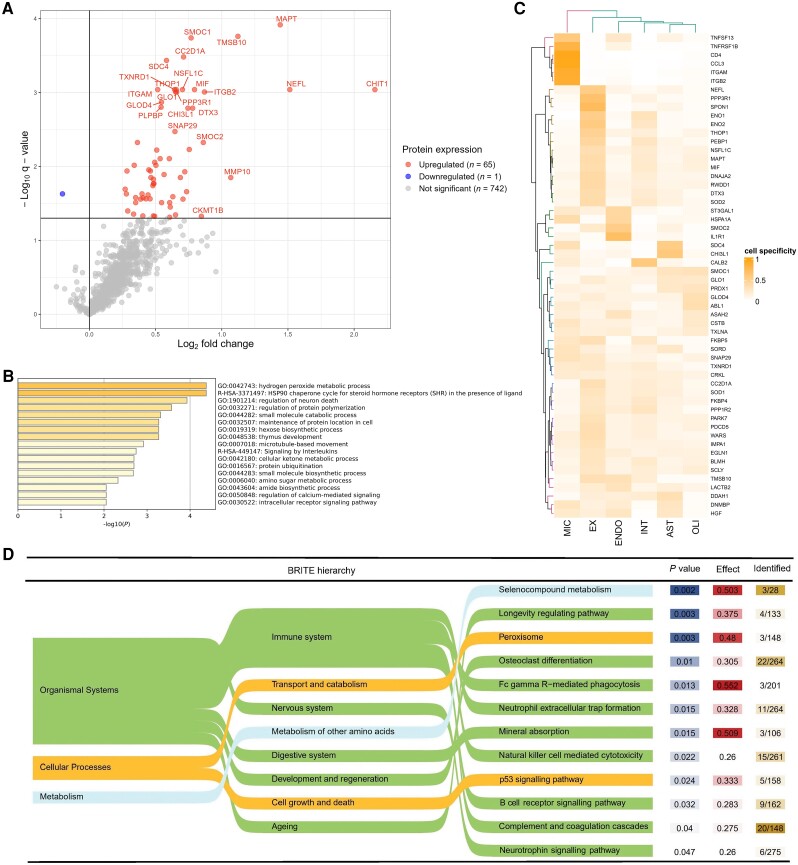
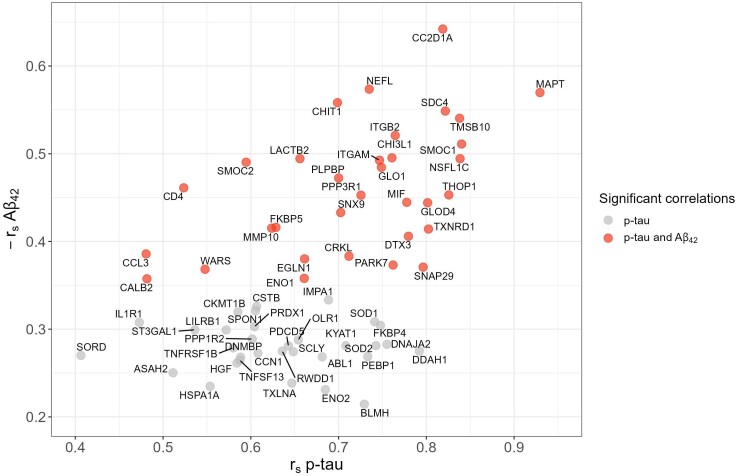
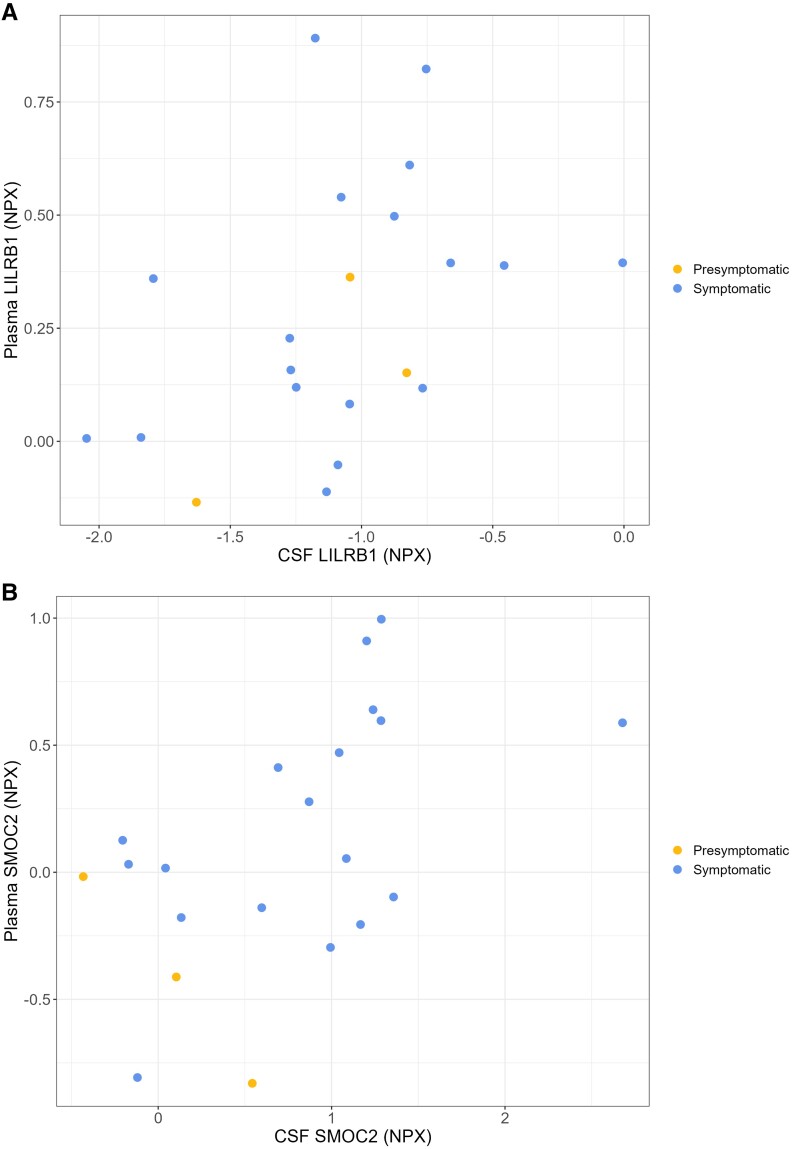
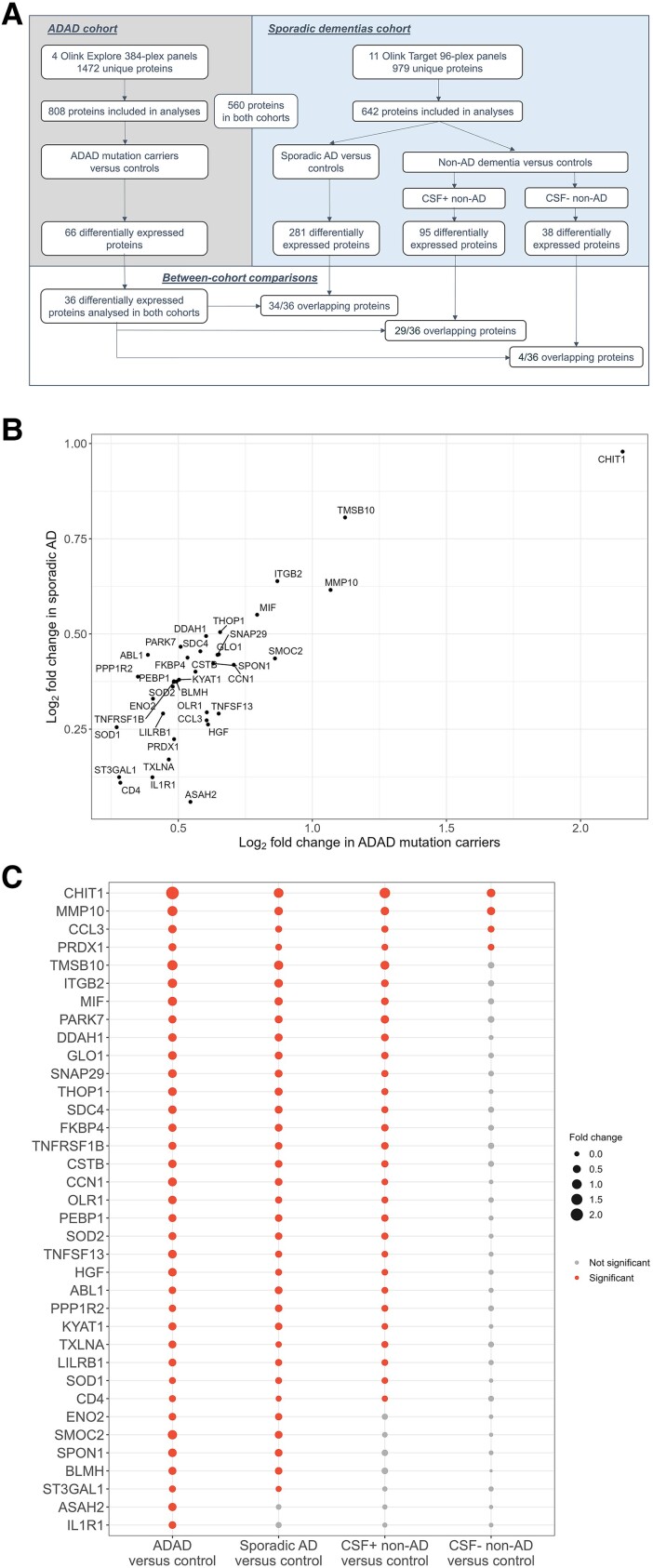
Autosomal dominant Alzheimer's disease (ADAD) offers a unique opportunity to study pathophysiological changes in a relatively young population with few comorbidities. A comprehensive investigation of proteome changes occurring in ADAD could provide valuable insights into AD-related biological mechanisms and uncover novel biomarkers and therapeutic targets. Furthermore, ADAD might serve as a model for sporadic AD, but in-depth proteome comparisons are lacking. We aimed to identify dysregulated CSF proteins in ADAD and determine the degree of overlap with sporadic AD. We measured 1472 proteins in CSF of PSEN1 or APP mutation carriers (n = 22) and age- and sex-matched controls (n = 20) from the Amsterdam Dementia Cohort using proximity extension-based immunoassays (PEA). We compared protein abundance between groups with two-sided t-tests and identified enriched biological pathways. Using the same protein panels in paired plasma samples, we investigated correlations between CSF proteins and their plasma counterparts. Finally, we compared our results with recently published PEA data from an international cohort of sporadic AD (n = 230) and non-AD dementias (n = 301). All statistical analyses were false discovery rate-corrected. We detected 66 differentially abundant CSF proteins (65 increased, 1 decreased) in ADAD compared to controls (q < 0.05). The most strongly upregulated proteins (fold change >1.8) were related to immunity (CHIT1, ITGB2, SMOC2), cytoskeletal structure (MAPT, NEFL) and tissue remodelling (TMSB10, MMP-10). Significant CSF-plasma correlations were found for the upregulated proteins SMOC2 and LILR1B. Of the 66 differentially expressed proteins, 36 had been measured previously in the sporadic dementias cohort, 34 of which (94%) were also significantly upregulated in sporadic AD, with a strong correlation between the fold changes of these proteins in both cohorts (rs = 0.730, P < 0.001). Twenty-nine of the 36 proteins (81%) were also upregulated among non-AD patients with suspected AD co-pathology. This CSF proteomics study demonstrates substantial biochemical similarities between ADAD and sporadic AD, suggesting involvement of the same biological processes. Besides known AD-related proteins, we identified several relatively novel proteins, such as TMSB10, MMP-10 and SMOC2, which have potential as novel biomarkers. With shared pathophysiological CSF changes, ADAD study findings might be translatable to sporadic AD, which could greatly expedite therapy development.
Keywords: dementia; familial; genetic; olink; proteome.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
A.W.L. has performed contract research with Axovant, EIP Pharma and Combinostics. All funding is paid to her institution. W.M.F. has performed contract research for Biogen MA Inc. and Boehringer Ingelheim, and has been an invited speaker at Boehringer Ingelheim, Biogen MA Inc, Danone, Eisai, WebMD Neurology (Medscape), NovoNordisk, Springer Healthcare and the European Brain Council. W.M.F. is consultant to Oxford Health Policy Forum CIC, Roche and Biogen MA Inc. She participated in the advisory boards of Biogen MA Inc., Roche and Eli Lilly. All funding is paid to her institution. W.M.F. is a member of the steering committee of PAVE and Think Brain Health. W.M.F. was associate editor of Alzheimer, Research & Therapy in 2020/2021 and is an associate editor at
Figures
References
-
- Lippa CF, Saunders AM, Smith TW, et al. Familial and sporadic Alzheimer’s disease: neuropathology cannot exclude a final common pathway. Neurology. 1996;46:406–412. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
