

doi: 10.1038/s41587-023-01831-7.
Epub 2023 Jun 22.
Single-cell m6A mapping in vivo using picoMeRIP-seq
Affiliations
- PMID: 37349523
- PMCID: PMC10739642
- DOI: 10.1038/s41587-023-01831-7
Item in Clipboard
Single-cell m6A mapping in vivo using picoMeRIP-seq
Nat Biotechnol.
2024 Apr.
Abstract
Current N6-methyladenosine (m6A) mapping methods need large amounts of RNA or are limited to cultured cells. Through optimized sample recovery and signal-to-noise ratio, we developed picogram-scale m6A RNA immunoprecipitation and sequencing (picoMeRIP-seq) for studying m6A in vivo in single cells and scarce cell types using standard laboratory equipment. We benchmark m6A mapping on titrations of poly(A) RNA and embryonic stem cells and in single zebrafish zygotes, mouse oocytes and embryos.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing financial interests.
Figures
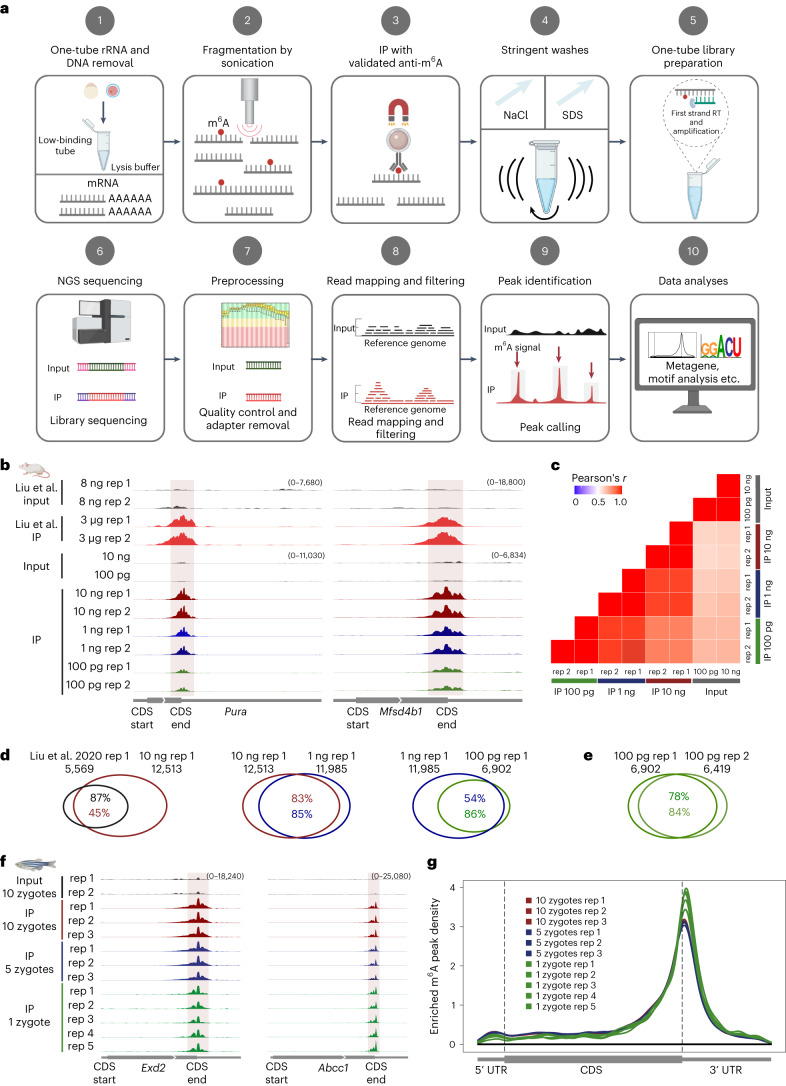
a, Schematic of the single-cell MeRIP–seq method and analysis pipeline; RT, reverse transcription; NGS, next-generation sequencing. Figure created with BioRender.com . b, Genome browser snapshots of two transcripts (transcript IDs: ENSMUST00000051301.5 for Pura and ENSMUST00000163705.2 for Mfsd4b1) harboring m6A enrichment near the stop codon. Tracks are shown for picoMeRIP–seq experiments with titration of the starting amount of poly(A)-selected RNA compared to published work starting with 3 μg of total RNA (Liu et al.). c, Transcriptome-wide correlation analyses (sequencing read coverage). d, Venn diagrams showing overlap of m6A peaks from picoMeRIP–seq experiments and comparison to published work starting with 3 μg of total RNA (Liu et al). e, Overlap of m6A peaks between two picoMeRIP–seq experiments from 100 pg of poly(A)-selected RNA. f, Genome browser snapshots of zebrafish zygote picoMeRIP–seq for two transcripts (transcript IDs: ENSDART00000111389 for Exd2 and ENSDART00000161897 for Abcc1) harboring m6A enrichment near the stop codon. g, Metagene profiles for zebrafish zygote picoMeRIP–seq showing the enrichment of m6A peaks along protein-coding gene transcripts.
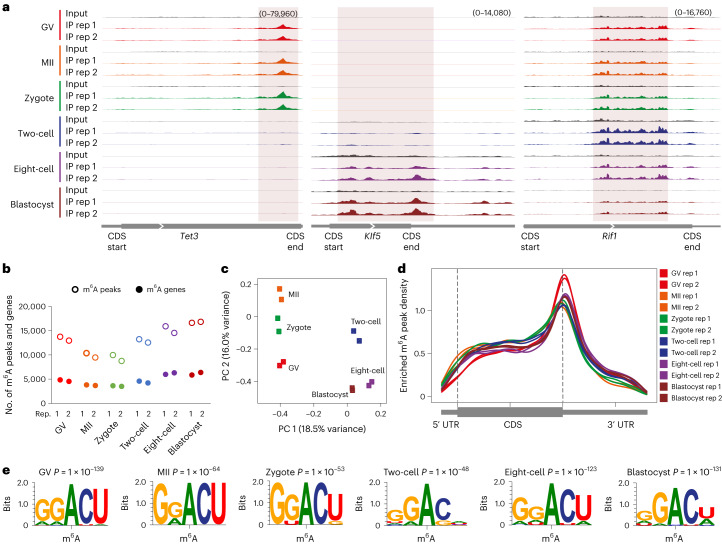
a, Genome browser snapshots showing three examples of transcripts with dynamic m6A enrichment during mouse oocyte and embryo development (transcript IDs: ENSMUST00000186548.6 for Tet3, ENSMUST00000005279.7 for Klf5 and ENSMUST00000112693.9 for Rif1). b, Number of m6A peaks and m6A-marked gene transcripts. c, PCA of m6A signal. d, Metagene profiles showing the enrichment of m6A peaks along protein-coding gene transcripts. e, Consensus motifs identified within m6A peaks identified in biological replicate 1.
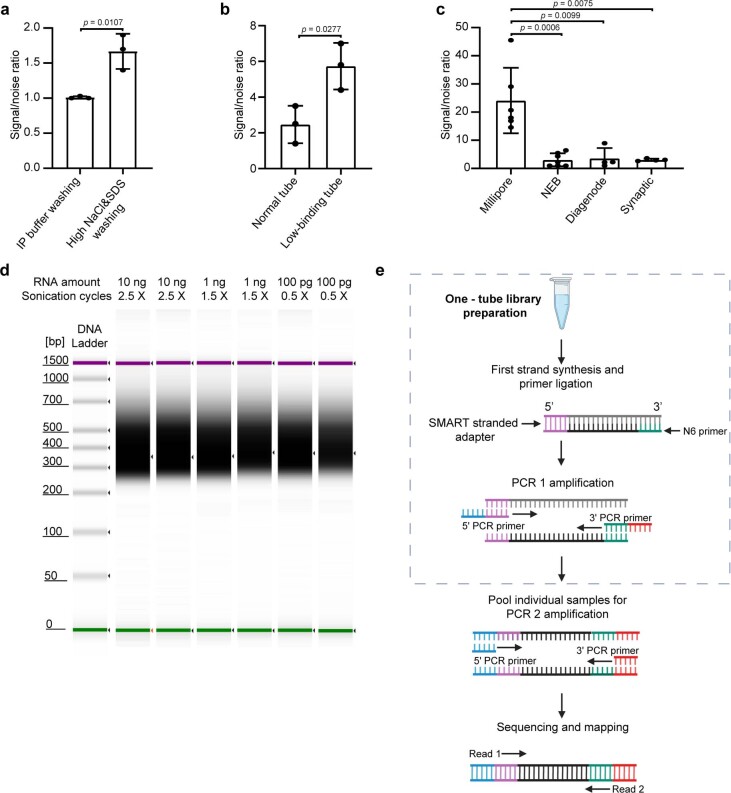
a, qPCR assessment of signal-to-noise ratio for the evaluation of the effect of increased stringency of the wash buffers. Mouse liver polyA selected RNA (10 ng) was used in these experiments. The previously validated m6A positive (Pdzd8) and negative (Rdh10) loci were used to calculate the signal-to-noise ratio (See Methods). Data are presented as mean ± standard deviation (SD), with n = 3 independent experiments for each experimental condition. Unpaired, two-tailed t-test was used. b, qPCR assessment of signal-to-noise ratio for the evaluation of the effect of low-binding tubes. Mouse liver polyA selected RNA (10 ng) was used in these experiments. Data are presented as mean ± SD values, with n = 3 independent experiments for each experimental condition. Unpaired, two-tailed t-test was used. c, qPCR assessment of signal-to-noise ratio for the optimized picoMeRIP method with different antibodies. Mouse liver polyA selected RNA (10 ng) was used in these experiments. Data are presented as mean ± SD values, with n = 6; 7; 4; 4 independent experiments for Millipore, NEB, Diagenode, Synaptic Systems antibodies, respectively. Unpaired, two-tailed t-test was used. d, Synthetic gel image from an Agilent 2100 Bioanalyzer electrophoreses run showing the size of sequencing libraries generated from picoMeRIP-seq of 10 ng, 1 ng and 100 pg mouse liver polyA selected RNA. Library preparation adds 139 bp to the size of the immunoprecipitated RNA fragments. The top line indicates the applied number of sonication cycles with the Hielscher UP100H sonicator. Each cycle is 30 seconds sonication plus 30 seconds on ice. e, Tailored SMART library preparation with a single-tube protocol to include as much as possible of the immunoprecipitated RNA. The SMART scN6 Primer (green) random primers allows the generation of cDNA from all immunoprecipitated RNA fragments. After the reverse transcription, the SMART Stranded Adapter (pink) will be linked with the cDNA product. Next, a first round of PCR amplification (PCR 1) adds full-length Illumina adapters, including barcodes. To minimize the number of samples to be processed downstream, individual samples can be pooled together for a second round of PCR amplification (PCR 2) using primers universal to all libraries. The final library is compatible with any Illumina sequencing platform. Created with BioRender.com.
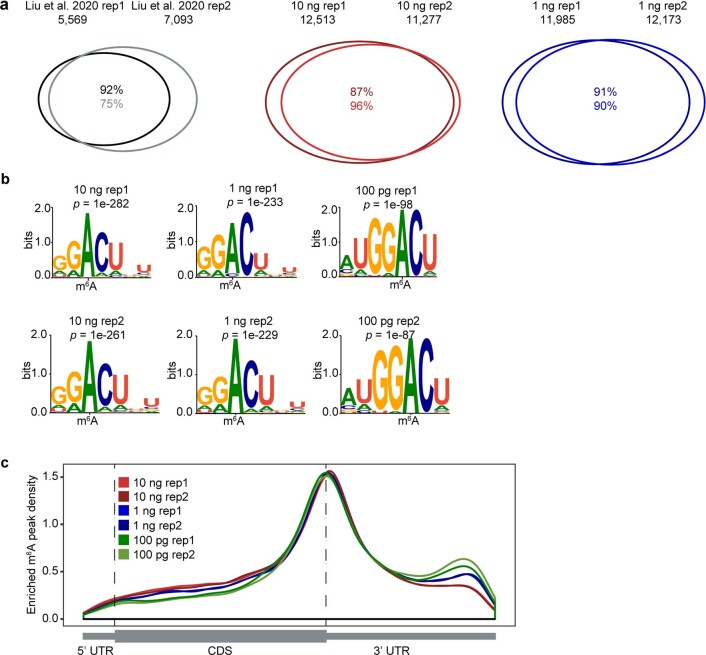
a, Venn diagrams showing overlap of m6A peaks from two replicates of published work starting with 3 µg total RNA (Liu et al), and for two replicates of picoMeRIP-seq experiments starting with either 10 ng or 1 ng of polyA RNA. b, Consensus motifs identified within m6A peaks from picoMeRIP-seq experiments with titration of the starting amount of polyA RNA. c, Metagene profiles showing the enrichment of m6A peaks along protein-coding gene transcripts.
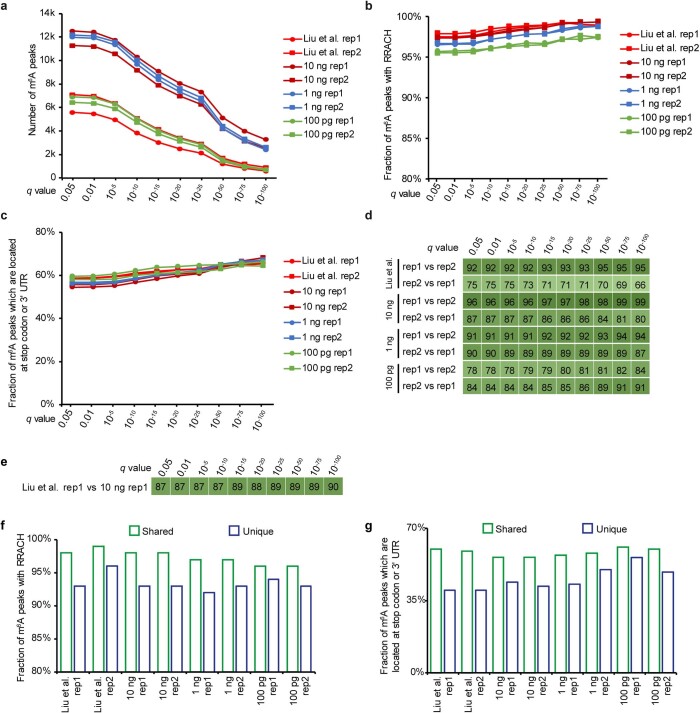
a, Number of m6A peaks. b, Fraction of peaks with RRACH motif. c, Fraction of peaks which are located at stop codon or 3’ UTR. d, Fraction of peaks overlapping between two biological replicates. e, Fraction of peaks overlapping between picoMeRIP-seq (10 ng, biological replicate 1) and published data (3 µg RNA, biological replicate 1). f, Fraction of peaks with RRACH motif for the peaks that are identified in both biological replicates (shared) and the peaks that are only supported by one replicate (unique), respectively. g, Fraction of peaks that are located at stop codon or 3’ UTR for the shared peaks and unique peaks, respectively.
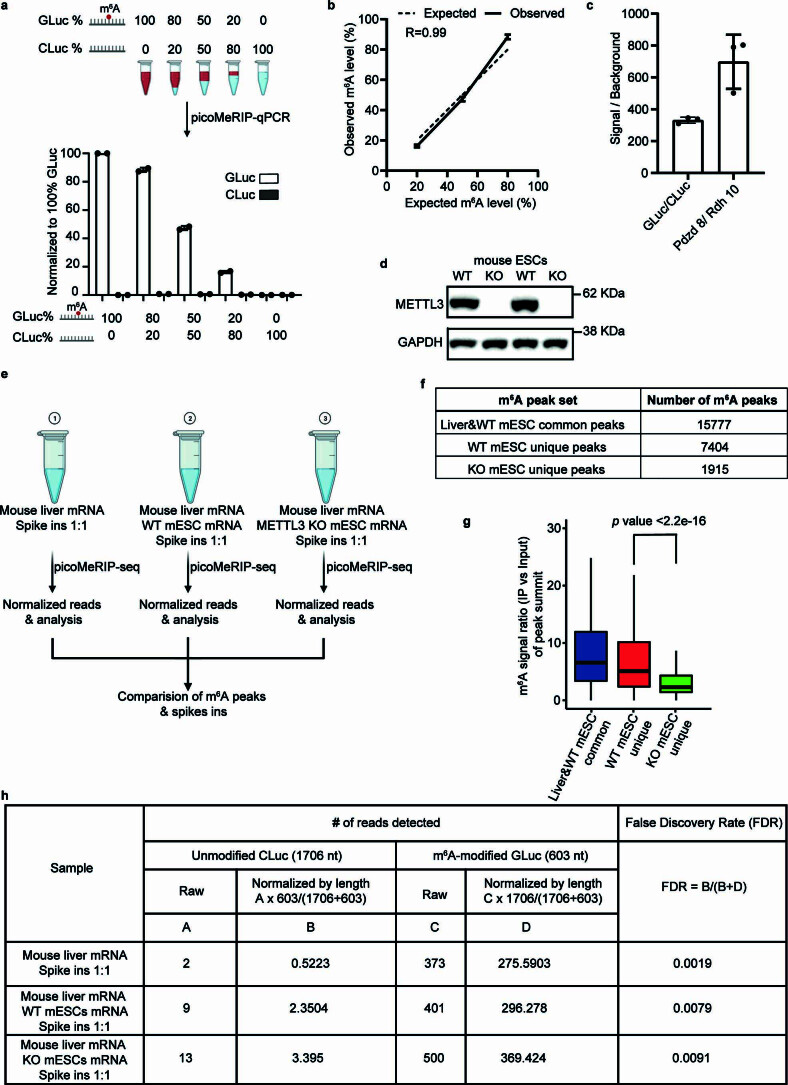
a, Top panel, schematic illustration of experimental setup for titration of two control RNAs, m6A-modified GLuc, and unmodified CLuc, for picoMeRIP and qPCR assessment. Bottom panel, quantified immunoprecipitation levels of m6A-modified GLuc, and unmodified CLuc, after picoMeRIP. picoMeRIP signal was normalized to 100% GLuc. Data are presented as mean ± SD values, with n = 2 independent experiments for each experimental condition. Created with BioRender.com. b, Experimentally observed m6A levels of m6A-modified control RNA as compared to the expected ones based on the titration experiment described in panel a. Data are presented as mean ± SD values, with n = 2 independent experiments for each experimental condition. R is the correlation coefficient in regression analysis. c, qPCR assessment of picoMeRIP m6A signal over background with two control RNAs used as spike-ins, m6A-modified GLuc, and unmodified CLuc, and for the previously validated m6A positive (Pdzd8) and negative (Rdh10) liver transcripts. Data are presented as mean ± SD values, with n = 3 independent experiments for each experimental condition. d, Western blot of METTL3 and GAPDH for WT and Mettl3 deficient (“knock-out” (KO)) mES cell lines. GAPDH was used as loading control. Two independent western blot experiments were performed with similar results. e, Schematic illustration of experimental setup to compare number of m6A peaks and peak signal strength of picoMeRIP-seq from WT and Mettl3 deficient (KO) mES cells, including spiked-in control RNAs. Mouse liver mRNA and mES cell mRNA were added in a 1:1 ratio. Two control RNAs were used as spike-ins, m6A-modified GLuc, and unmodified CLuc, and added at a 1:1 ratio. Created with BioRender.com. f, Number of picoMeRIP-seq m6A peaks uniquely identified in WT and Mettl3 deficient (KO) mES cells, and co-identified in mouse liver and WT mES cells. g, Box plots of m6A signal for comparison of picoMeRIP-seq peaks uniquely identified in WT and Mettl3 deficient (KO) mES cells. Middle vertical line (bold) depicts the median, and the 25th percentile to 75th percentile is shown. Whiskers extend to the most extreme data point within 1.5 interquartile range of the quartiles. The p value was calculated using a Wilcoxon rank sum test (two-sided). The corresponding m6A peak numbers for each of three groups are shown in panel f. h, Assessment of false discovery rate of picoMeRIP-seq by spike-in m6A-modified GLuc, and unmodified Cluc control RNAs. Source data
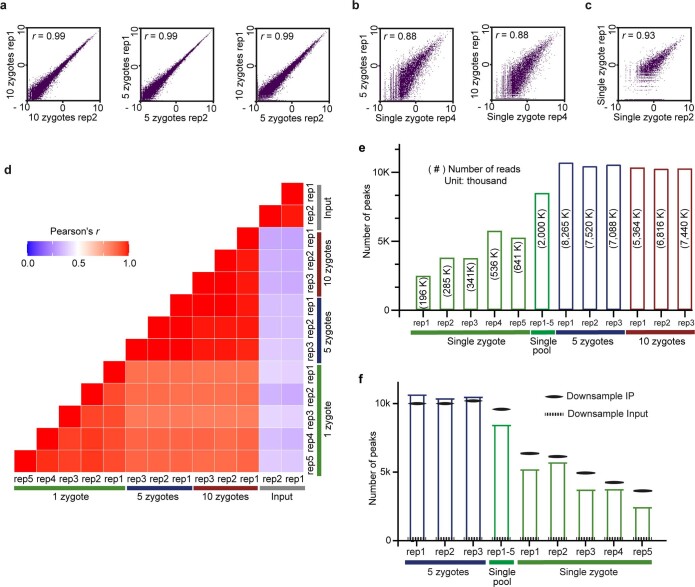
a, b, c, Scatter plots showing the correlation of picoMeRIP-seq experiments for (a) pooled zygotes; (b) pooled zygotes and single zygote; and (c) single zygote. The scatter plots are plotted using the natural log of read coverage (RPKM + 1). r, Pearson correlation coefficient. d, Heatmap comparing picoMeRIP-seq experiments from pooled zygotes and single zygote. r, Pearson correlation coefficient. e, Number of m6A peaks. Peak detection when combining the data from five single-zygote experiments is also shown. The numbers of uniquely mapped, deduplicated, non-rRNA-derived sequencing reads are shown inside the bars within parentheses. f, Number of m6A peaks called from different picoMeRIP-seq experiments from pools of zygotes and for single zygotes shown as full columns, as reference. Number of m6A peaks called from randomly down-sampled data from a picoMeRIP-seq experiments from a pool of ten zygotes is indicated with solid black lines. Down-sampling was performed to produce equal reads numbers as that originally yielded from each of the indicated experiments. The numbers of detected peaks from single-zygote picoMeRIP-seq is close to the expected maximum as estimated by peak calling from randomly down-sampled data from pools of ten zygotes. As a control, peak calling was also performed from randomly down-sampled data from input.
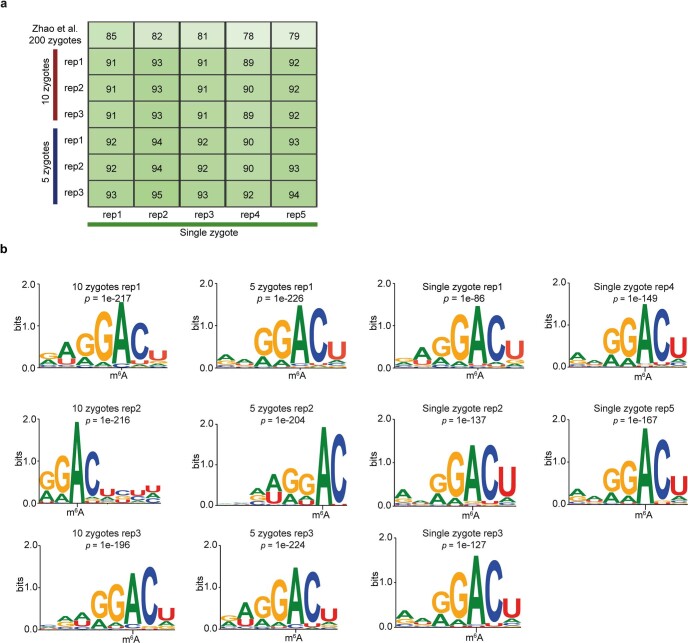
a, Fraction of overlap of m6A peaks between single-zygote picoMeRIP-seq experiments and picoMeRIP-seq from pools of zygotes. The topmost row is the fraction of overlap of m6A transcripts with previously published work (Zhao et al, GEO accession: GSM2088167 and GSM2088177 ). b, Consensus motifs identified within m6A peaks from single-zygote picoMeRIP-seq experiments and for picoMeRIP-seq from pools of zygotes.
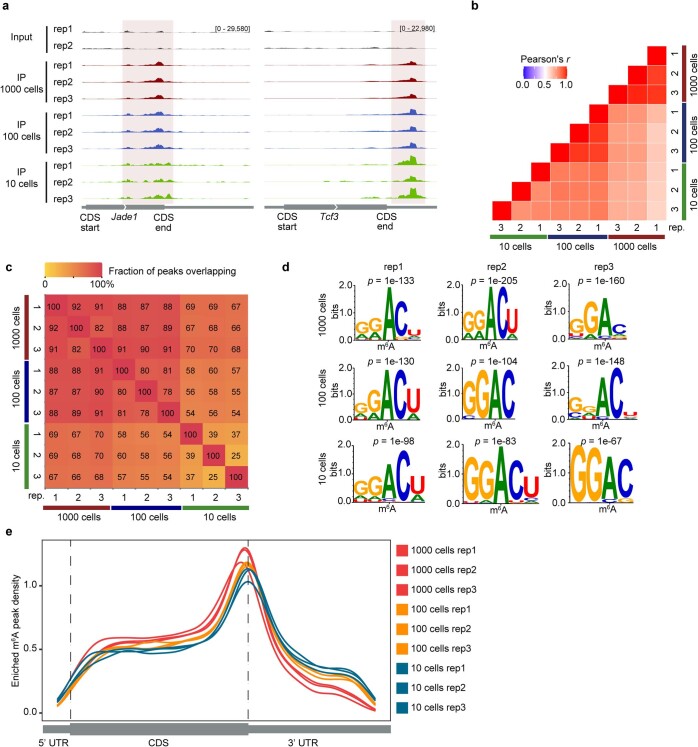
a, Genome browser snap shots of two transcripts with m6A enrichment (transcript ID: ENSMUST00000026865.14 for Jade1, ENSMUST00000105344.7 for Tcf3). b, Transcriptome-wide correlation analyses (sequencing read coverage) between picoMeRIP-seq experiments from 1,000, 100 and 10 mouse ES cells. c, Heatmap showing fraction of overlap of m6A peaks between picoMeRIP-seq experiments with 1,000, 100 and 10 mouse ES cells. Peaks identified in the samples indicated at the bottom of the plot were used as the reference when calculating fraction of overlap of peaks from samples indicated at the left side of the plot. d, Consensus motifs identified within m6A peaks from picoMeRIP-seq experiments with 1,000, 100 and 10 mouse ES cells. e, Metagene profiles showing the enrichment of m6A peaks along protein-coding gene transcripts for 1,000, 100 and 10 mouse ES cells.
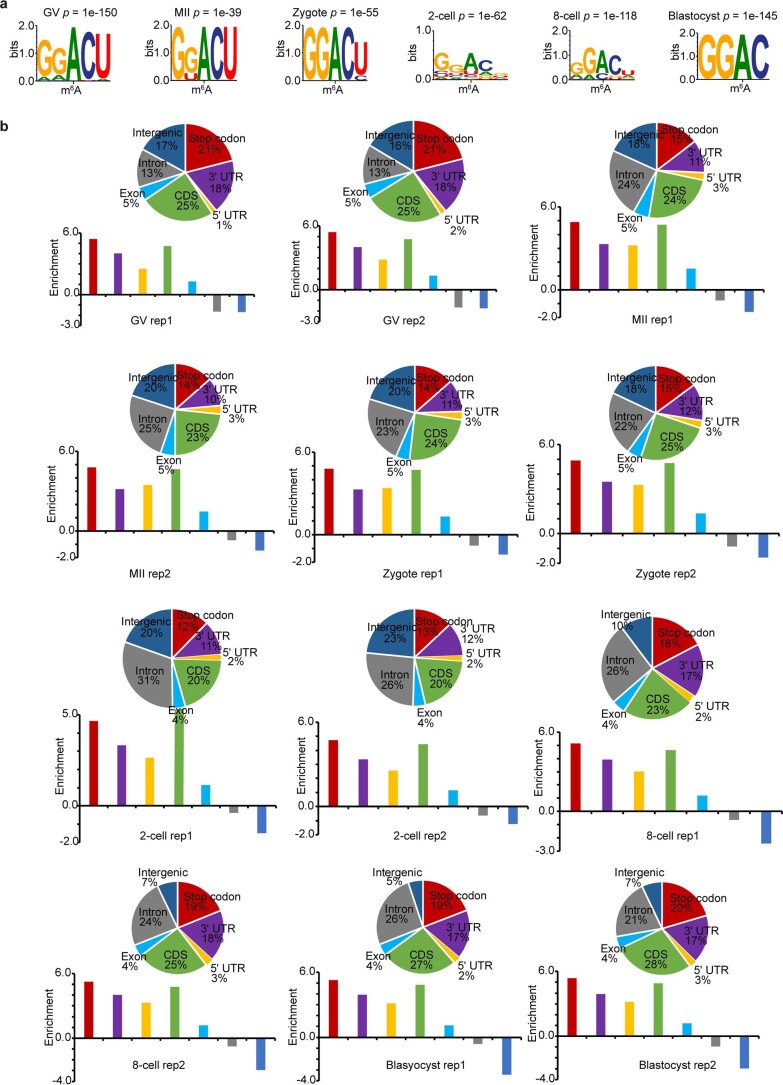
a, Consensus motifs identified within m6A peaks of biological replicate 2. b, Top, pie charts show genomic annotation of m6A peaks. Bottom, bar plots show the peak enrichment score, which was calculated as the log2 ratio of the observed over expected peak numbers.
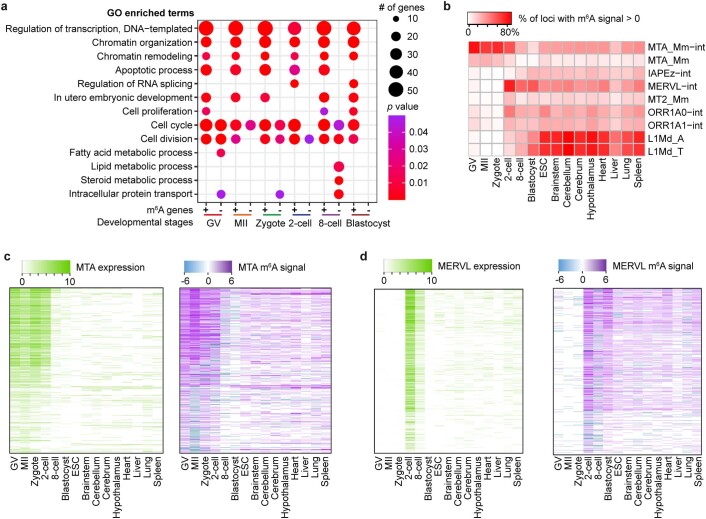
a, Gene Ontology (GO) analysis for m6A-marked transcripts (+) and for transcripts not marked by m6A (-). The modified Fisher exact p values were (that is EASE score) reported by DAVID (see https://david.ncifcrf.gov/helps/functional_annotation.html ). b, Enrichment of m6A in transcripts from selected retrotransposon subfamilies shown for mouse oocytes, embryos, ES cells (ESCs) and eight mouse tissues. MeRIP-seq data for mouse tissues are from Liu et al.. c, d, Expression and m6A signal profiles across the internal sequences of MTA (c); and MERVL (d). The expression is RPKM calculated by deepTools (see Methods).
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
