

Strain dropouts reveal interactions that govern the metabolic output of the gut microbiome
- PMID: 37352836
- PMCID: PMC10299816
- DOI: 10.1016/j.cell.2023.05.037
Strain dropouts reveal interactions that govern the metabolic output of the gut microbiome
Abstract
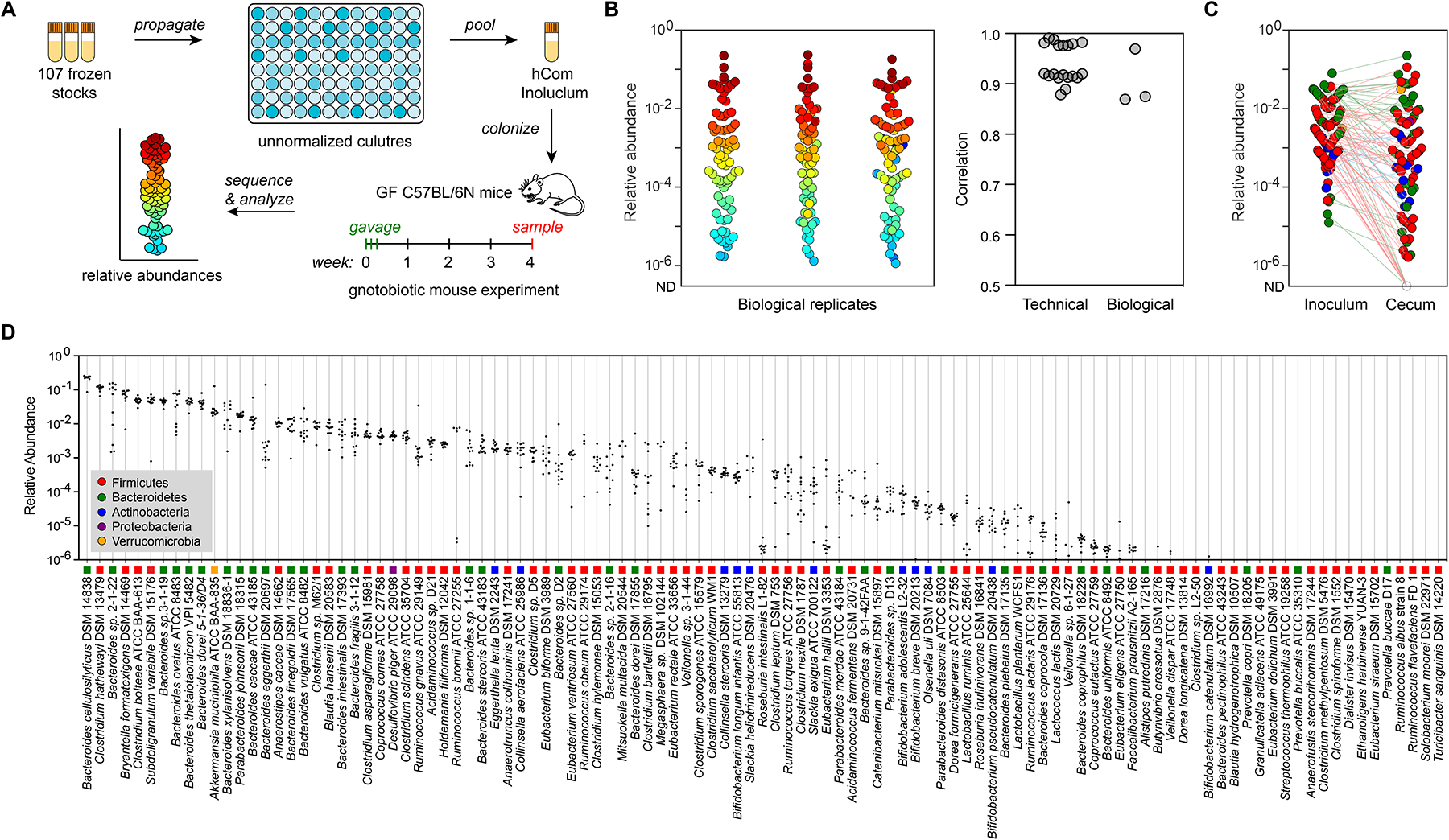
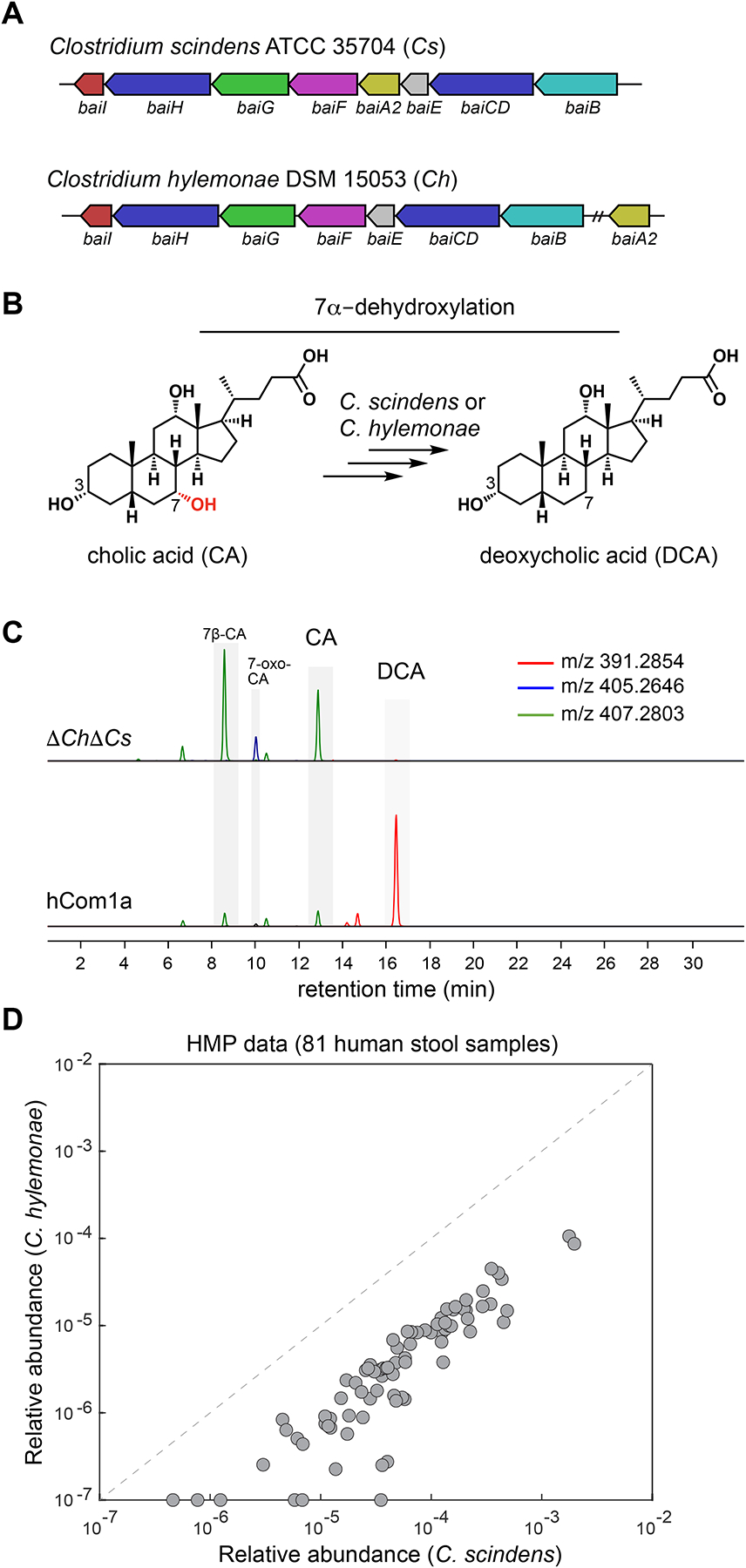
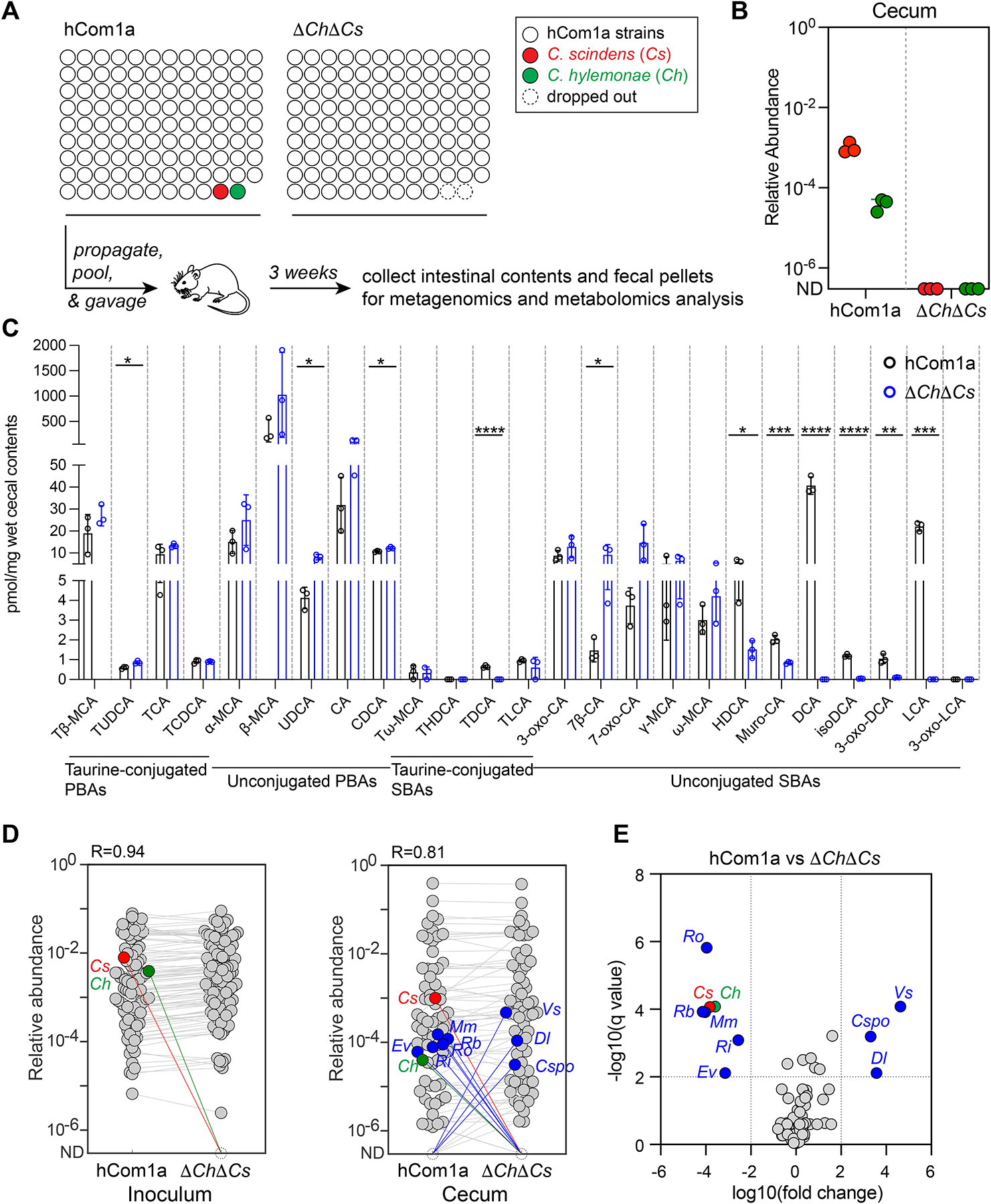
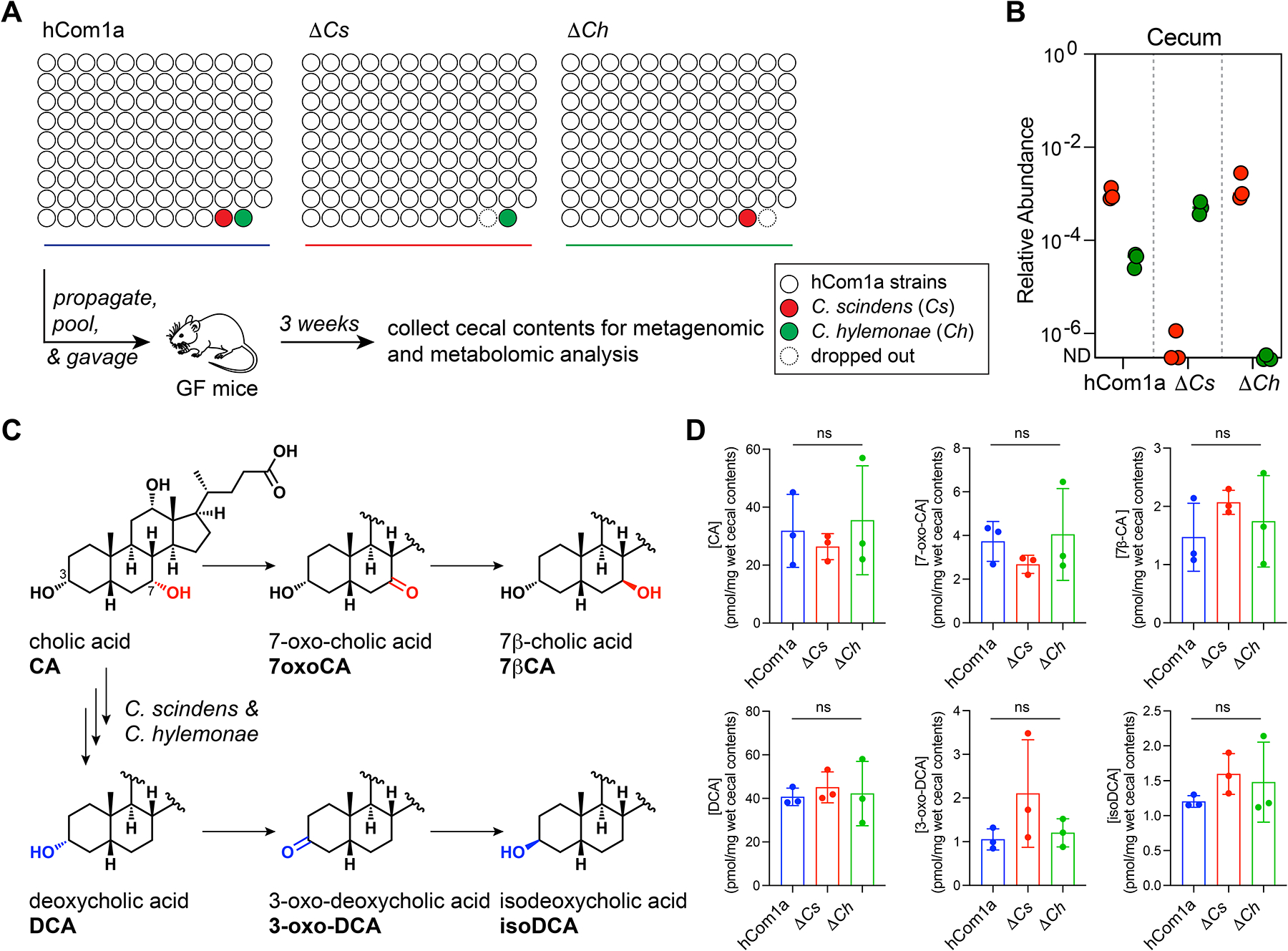
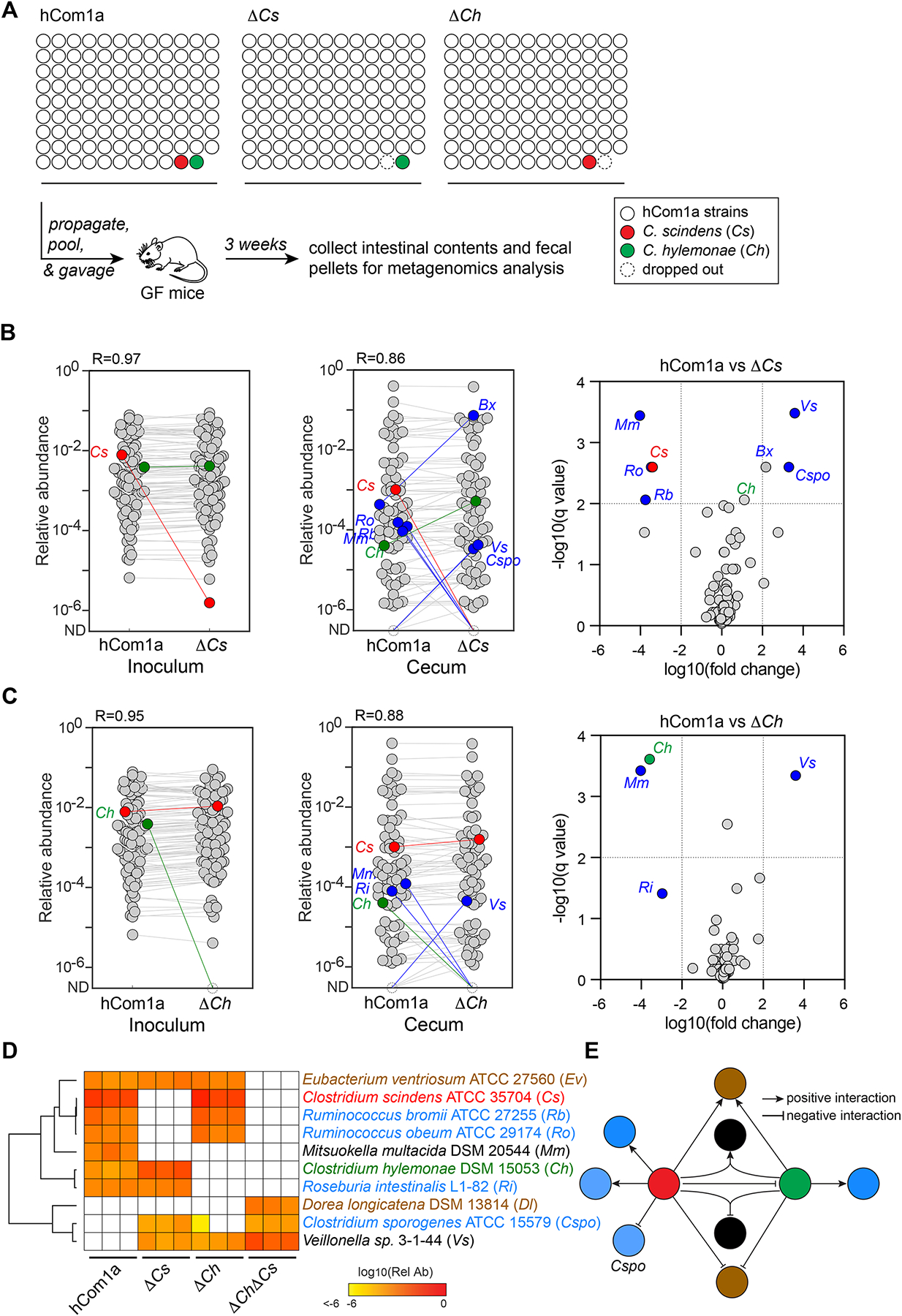
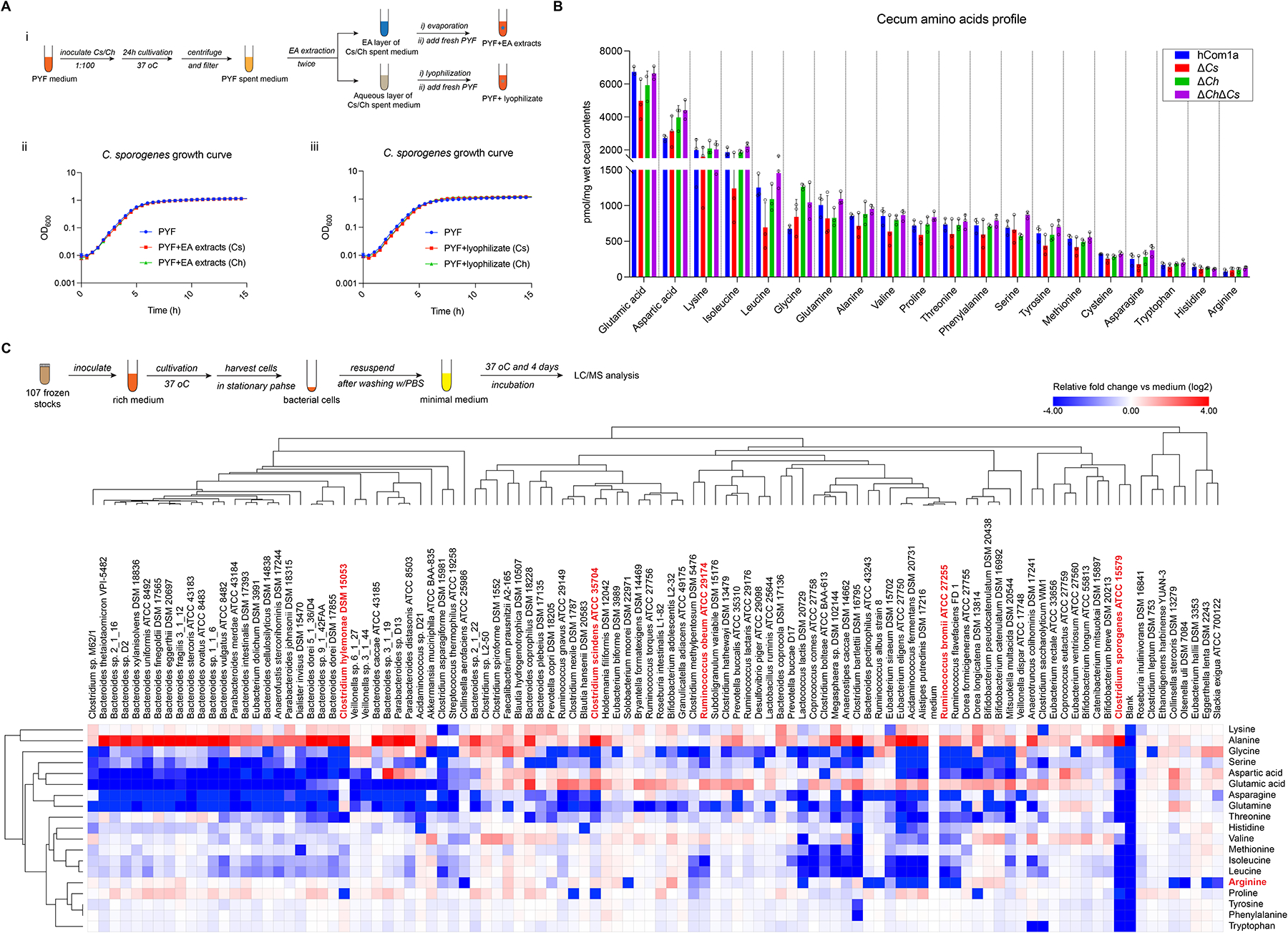
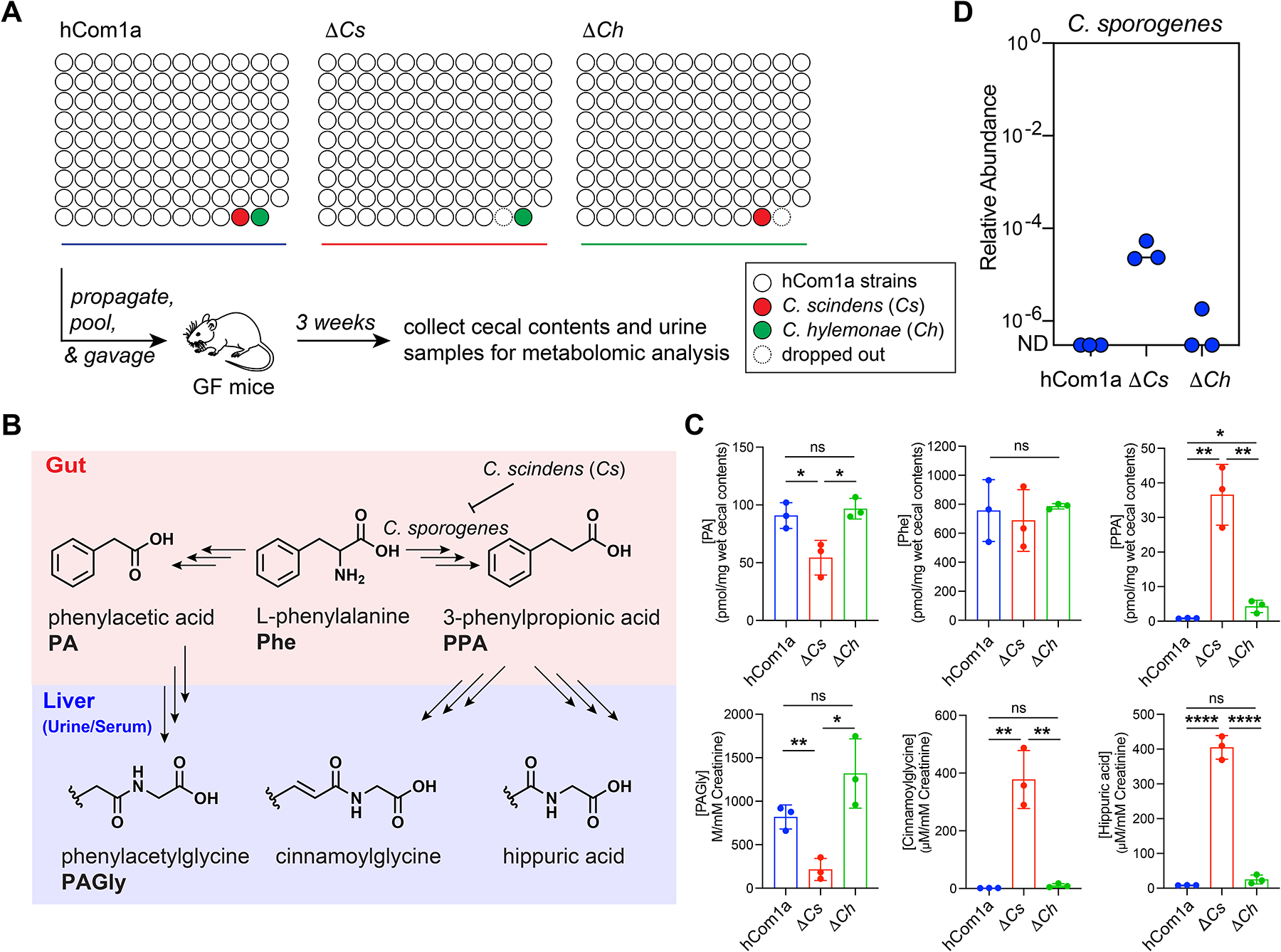
The gut microbiome is complex, raising questions about the role of individual strains in the community. Here, we address this question by constructing variants of a complex defined community in which we eliminate strains that occupy the bile acid 7α-dehydroxylation niche. Omitting Clostridium scindens (Cs) and Clostridium hylemonae (Ch) eliminates secondary bile acid production and reshapes the community in a highly specific manner: eight strains change in relative abundance by >100-fold. In single-strain dropout communities, Cs and Ch reach the same relative abundance and dehydroxylate bile acids to a similar extent. However, Clostridium sporogenes increases >1,000-fold in the ΔCs but not ΔCh dropout, reshaping the pool of microbiome-derived phenylalanine metabolites. Thus, strains that are functionally redundant within a niche can have widely varying impacts outside the niche, and a strain swap can ripple through the community in an unpredictable manner, resulting in a large impact on an unrelated community-level phenotype.
Keywords: ecology; metabolism; microbiome.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests Stanford University and the Chan Zuckerberg Biohub have patents pending for microbiome technologies on which the authors are co-inventors. M.A.F. is a co-founder and director of Federation Bio and Kelonia, a co-founder of Revolution Medicines, an Innovation Partner at the Column Group, and a member of the scientific advisory board of NGM Bio.
Figures
References
-
- Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang C, Magrini V, Wilson RK, et al. (2009). Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc. Natl. Acad. Sci 106, 5859–5864. 10.1073/pnas.0901529106. - DOI - PMC - PubMed
-
- Studer N, Desharnais L, Beutler M, Brugiroux S, Terrazos MA, Menin L, Schürch CM, McCoy KD, Kuehne SA, Minton NP, et al. (2016). Functional Intestinal Bile Acid 7α-Dehydroxylation by Clostridium scindens Associated with Protection from Clostridium difficile Infection in a Gnotobiotic Mouse Model. Front. Cell. Infect. Microbiol 6, 1–15. 10.3389/fcimb.2016.00191. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
