

Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup
- PMID: 37352859
- PMCID: PMC10357475
- DOI: 10.1016/j.ajhg.2023.06.002
Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup
Abstract
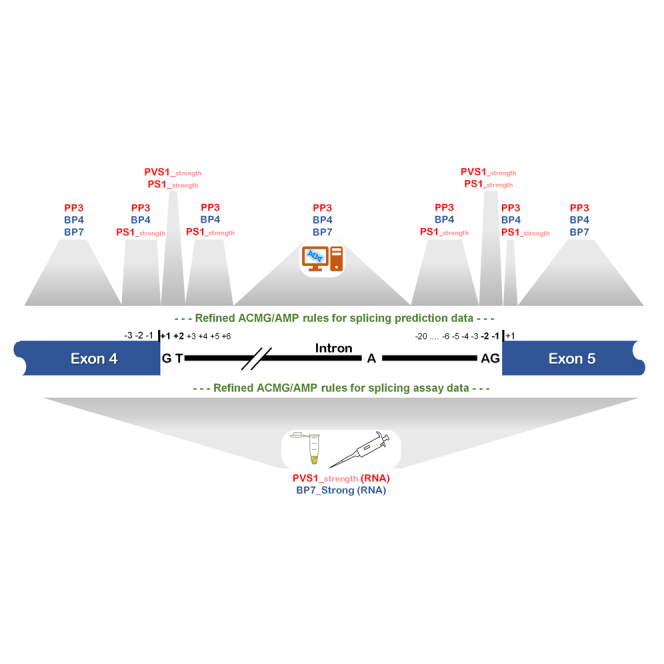
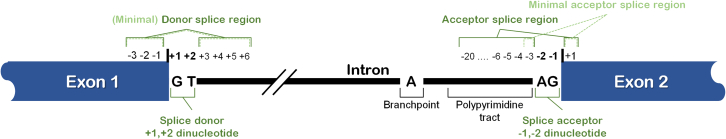
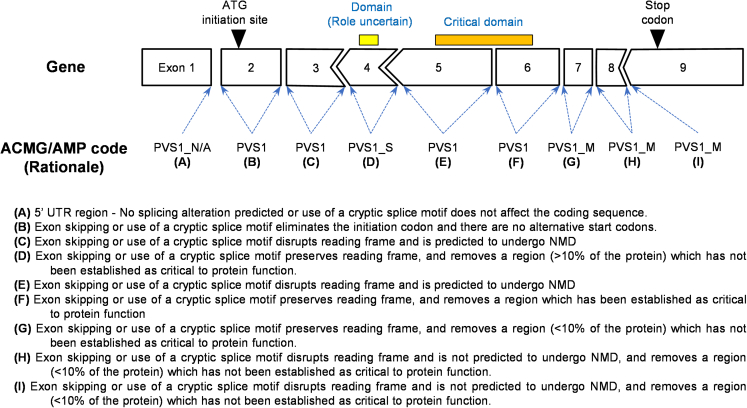
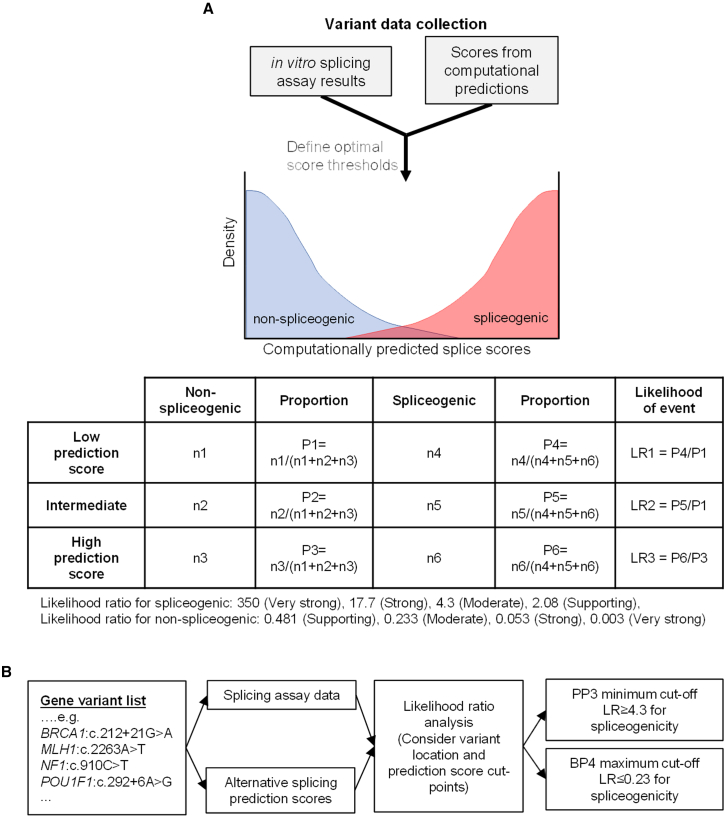
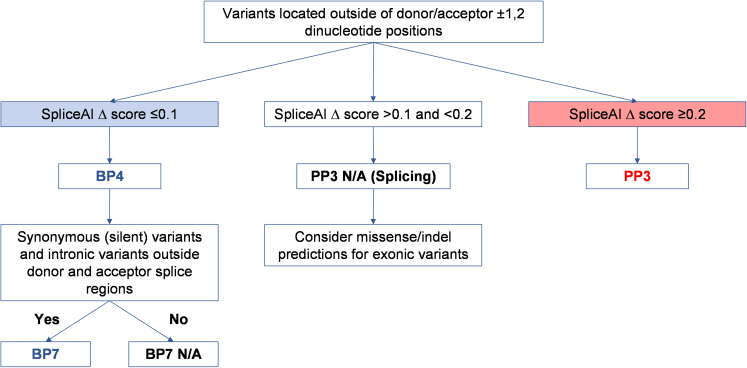
The American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) framework for classifying variants uses six evidence categories related to the splicing potential of variants: PVS1, PS3, PP3, BS3, BP4, and BP7. However, the lack of guidance on how to apply such codes has contributed to variation in the specifications developed by different Clinical Genome Resource (ClinGen) Variant Curation Expert Panels. The ClinGen Sequence Variant Interpretation Splicing Subgroup was established to refine recommendations for applying ACMG/AMP codes relating to splicing data and computational predictions. We utilized empirically derived splicing evidence to (1) determine the evidence weighting of splicing-related data and appropriate criteria code selection for general use, (2) outline a process for integrating splicing-related considerations when developing a gene-specific PVS1 decision tree, and (3) exemplify methodology to calibrate splice prediction tools. We propose repurposing the PVS1_Strength code to capture splicing assay data that provide experimental evidence for variants resulting in RNA transcript(s) with loss of function. Conversely, BP7 may be used to capture RNA results demonstrating no splicing impact for intronic and synonymous variants. We propose that the PS3/BS3 codes are applied only for well-established assays that measure functional impact not directly captured by RNA-splicing assays. We recommend the application of PS1 based on similarity of predicted RNA-splicing effects for a variant under assessment in comparison with a known pathogenic variant. The recommendations and approaches for consideration and evaluation of RNA-assay evidence described aim to help standardize variant pathogenicity classification processes when interpreting splicing-based evidence.
Keywords: ACMG/AMP codes; BP4; BP7; ClinGen; PP3; PS1; PVS1; RNA splicing; variant classification.
Copyright © 2023 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.L., L.M.V., S.H., H.Z., R.K., D.B.-B., A.C., A.T., and T.P. are employed by fee-for-service laboratories performing clinical sequencing services.
Figures

Update of
-
APPLICATION OF THE ACMG/AMP FRAMEWORK TO CAPTURE EVIDENCE RELEVANT TO PREDICTED AND OBSERVED IMPACT ON SPLICING: RECOMMENDATIONS FROM THE CLINGEN SVI SPLICING SUBGROUP.medRxiv [Preprint]. 2023 Feb 26:2023.02.24.23286431. doi: 10.1101/2023.02.24.23286431. medRxiv. 2023. Update in: Am J Hum Genet. 2023 Jul 6;110(7):1046-1067. doi: 10.1016/j.ajhg.2023.06.002. PMID: 36865205 Free PMC article. Updated. Preprint.
References
-
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. - DOI - PMC - PubMed
-
- Abou Tayoun A.N., Pesaran T., DiStefano M.T., Oza A., Rehm H.L., Biesecker L.G., Harrison S.M., ClinGen Sequence Variant Interpretation Working Group ClinGen SVI Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018;39:1517–1524. doi: 10.1002/humu.23626. - DOI - PMC - PubMed
-
- Brnich S.E., Abou Tayoun A.N., Couch F.J., Cutting G.R., Greenblatt M.S., Heinen C.D., Kanavy D.M., Luo X., McNulty S.M., Starita L.M., et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019;12:3. doi: 10.1186/s13073-019-0690-2. - DOI - PMC - PubMed
-
- Spurdle A.B., Greville-Heygate S., Antoniou A.C., Brown M., Burke L., de la Hoya M., Domchek S., Dörk T., Firth H.V., Monteiro A.N., et al. Towards controlled terminology for reporting germline cancer susceptibility variants: an ENIGMA report. J. Med. Genet. 2019;56:347–357. doi: 10.1136/jmedgenet-2018-105872. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
