

VarID2 quantifies gene expression noise dynamics and unveils functional heterogeneity of ageing hematopoietic stem cells
- PMID: 37353813
- PMCID: PMC10290360
- DOI: 10.1186/s13059-023-02974-1
VarID2 quantifies gene expression noise dynamics and unveils functional heterogeneity of ageing hematopoietic stem cells
Abstract
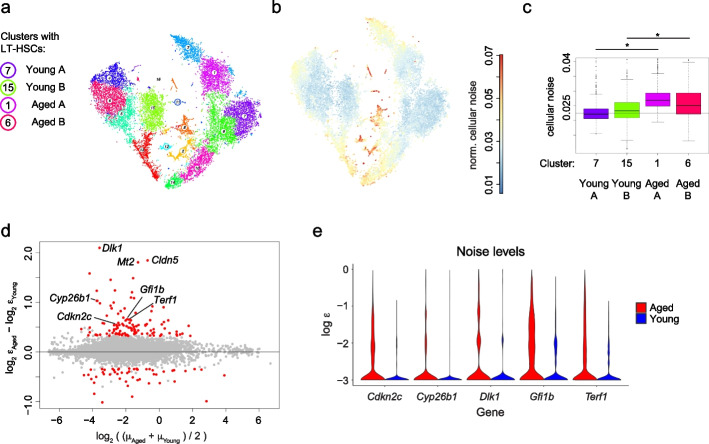
Variability of gene expression due to stochasticity of transcription or variation of extrinsic signals, termed biological noise, is a potential driving force of cellular differentiation. Utilizing single-cell RNA-sequencing, we develop VarID2 for the quantification of biological noise at single-cell resolution. VarID2 reveals enhanced nuclear versus cytoplasmic noise, and distinct regulatory modes stratified by correlation between noise, expression, and chromatin accessibility. Noise levels are minimal in murine hematopoietic stem cells (HSCs) and increase during differentiation and ageing. Differential noise identifies myeloid-biased Dlk1+ long-term HSCs in aged mice with enhanced quiescence and self-renewal capacity. VarID2 reveals noise dynamics invisible to conventional single-cell transcriptome analysis.
Keywords: Ageing; Cell sate variability; Gene expression noise; Hematopoietic stem cells; Machine learning; Mathematical modeling; Single-cell RNA sequencing; Stem cell differentiation.
© 2023. The Author(s).
Conflict of interest statement
DG serves on the scientific advisory board of Gordian Biotechnology.
Figures





References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
