

Reduced NR2F2 Expression in the Host Response to Infectious Bursal Disease Virus Infection Suppressed Viral Replication by Enhancing Type I Interferon Expression by Targeting SOCS5
- PMID: 37358466
- PMCID: PMC10373545
- DOI: 10.1128/jvi.00664-23
Reduced NR2F2 Expression in the Host Response to Infectious Bursal Disease Virus Infection Suppressed Viral Replication by Enhancing Type I Interferon Expression by Targeting SOCS5
Abstract
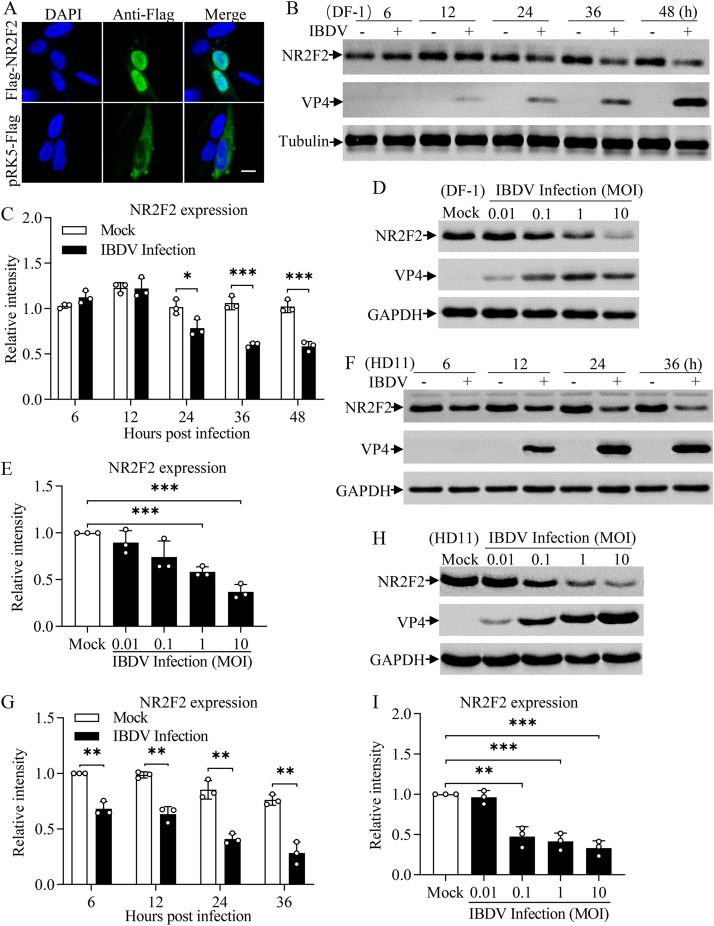
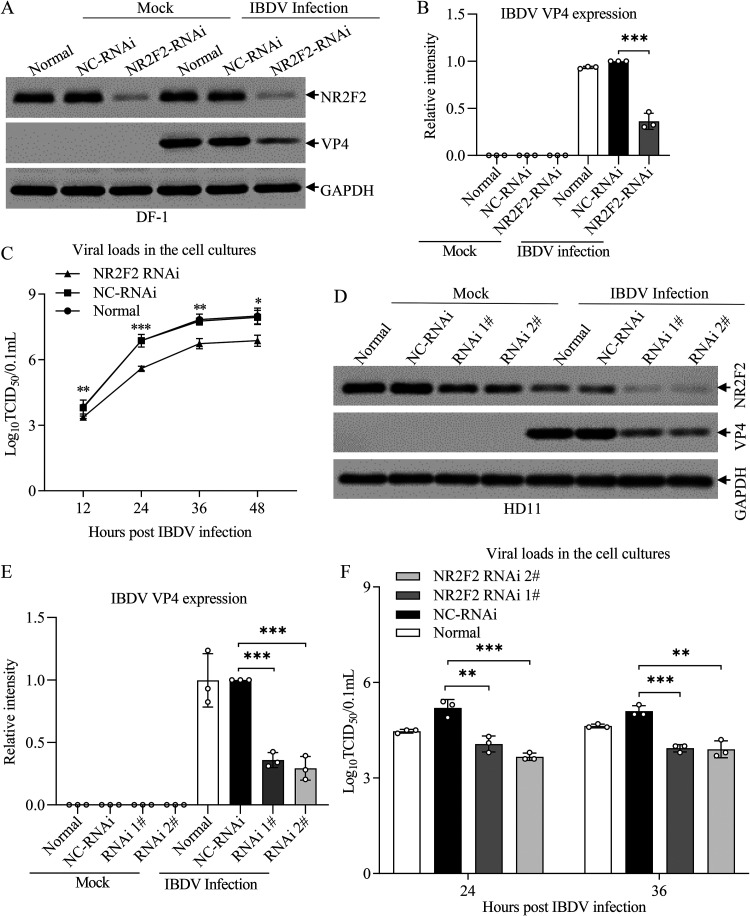
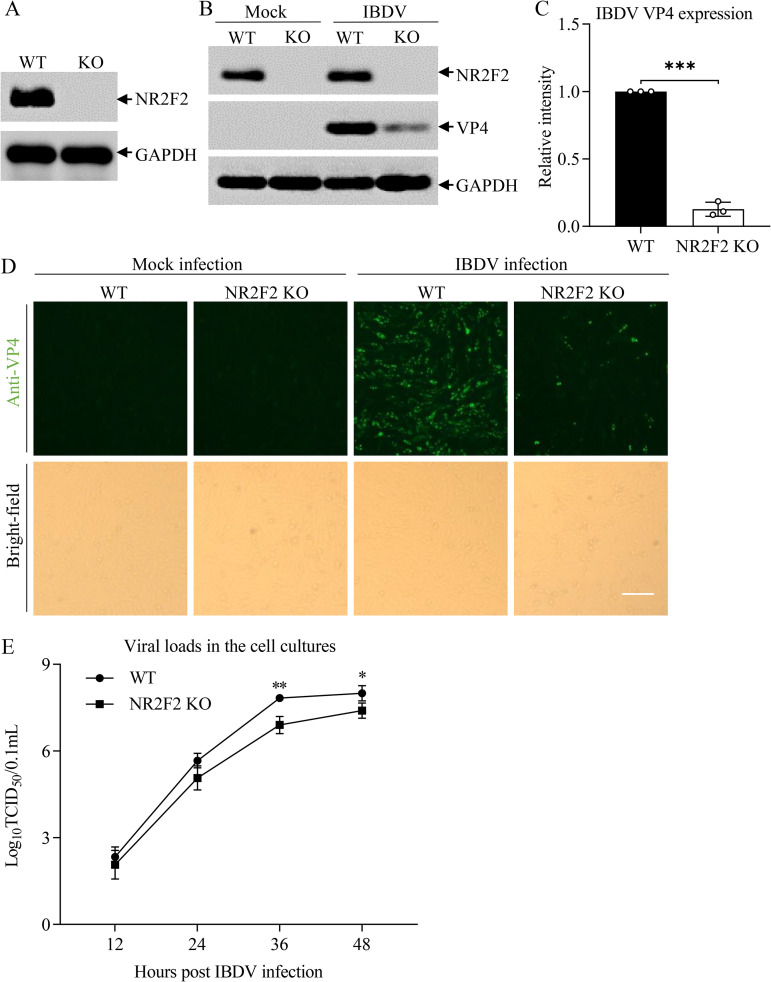
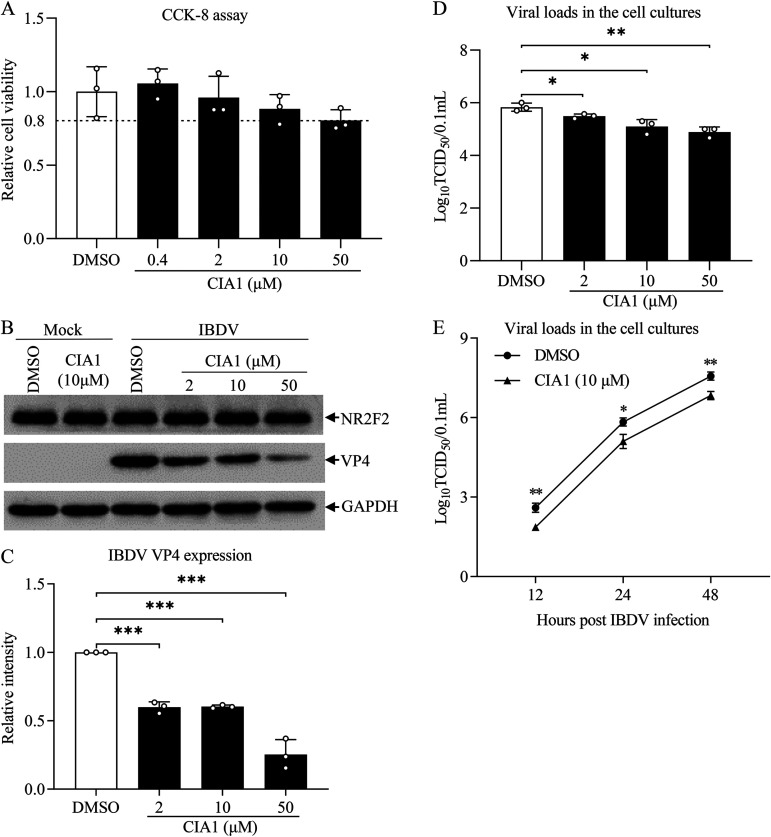
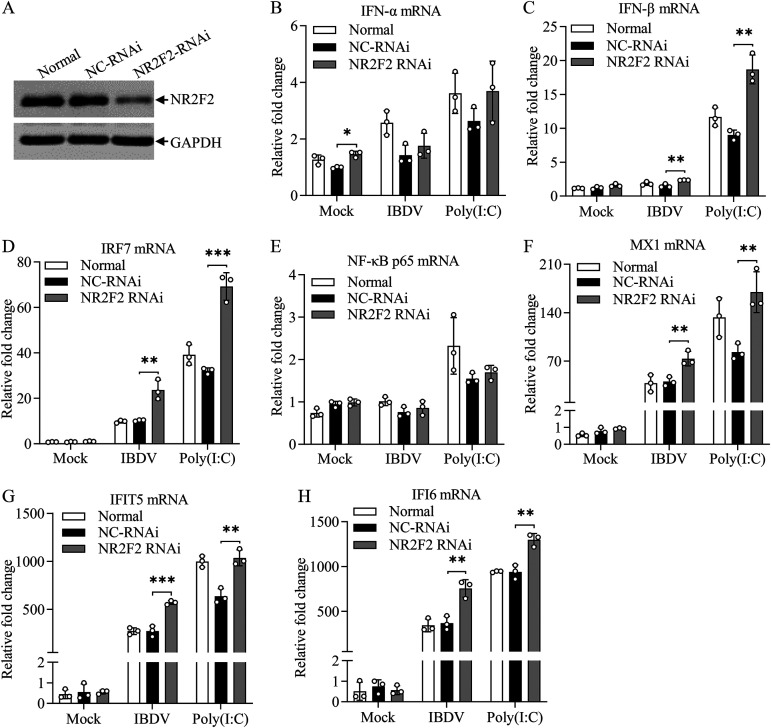
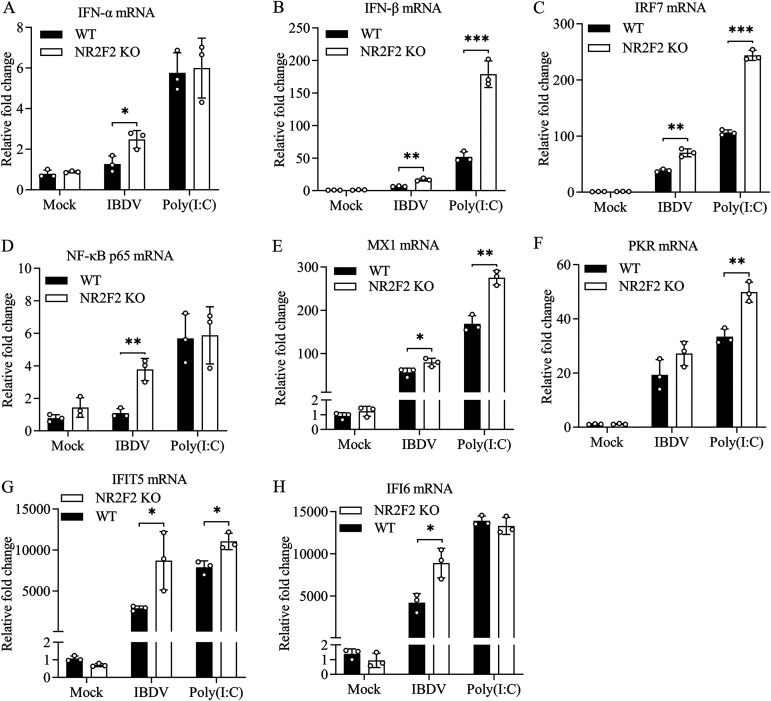
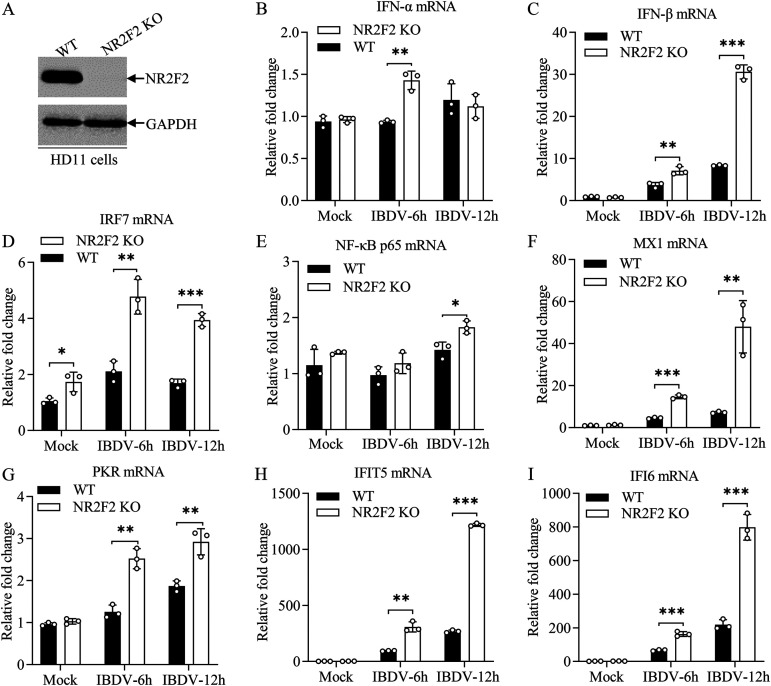
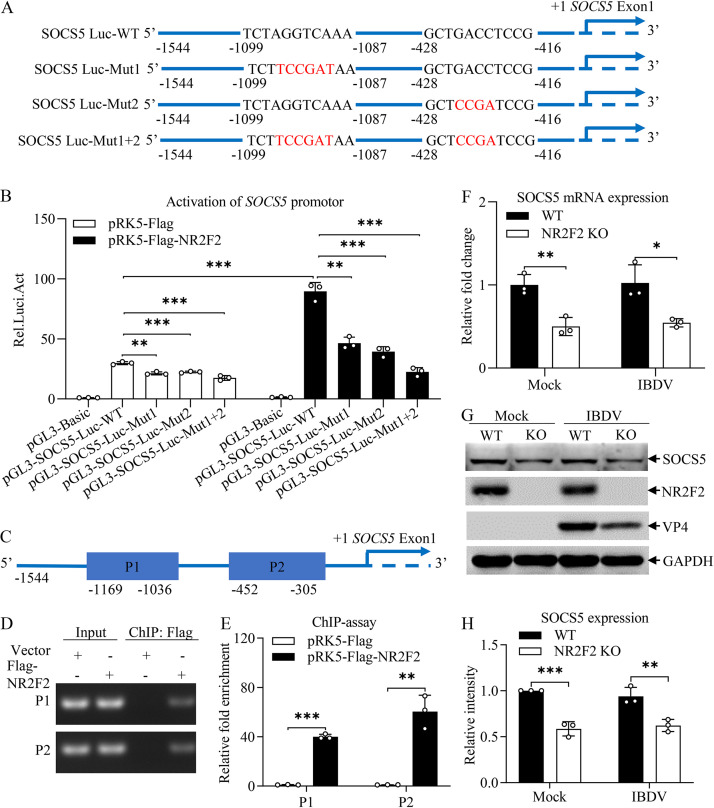
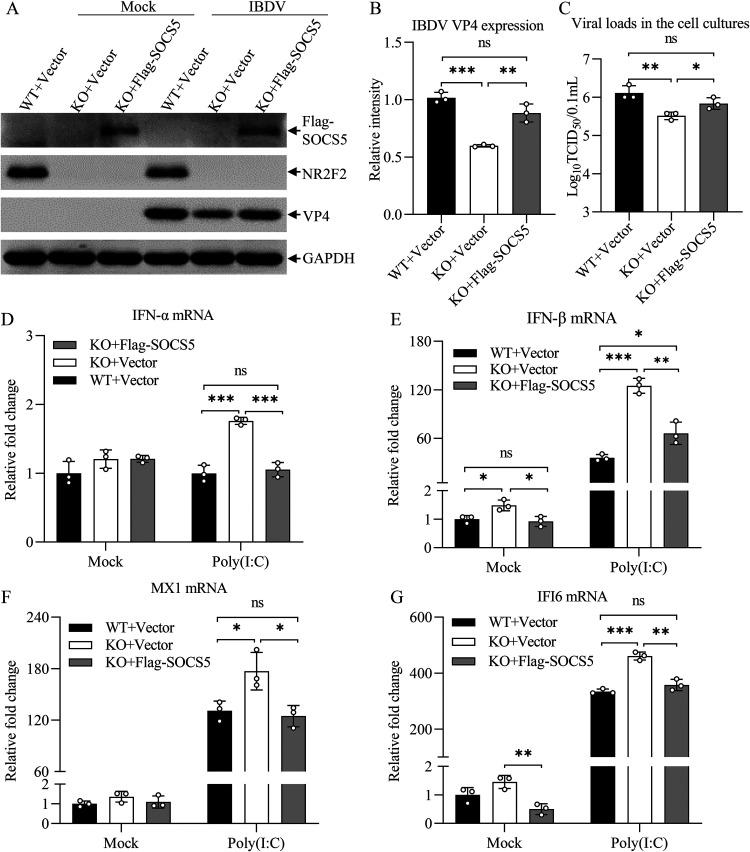
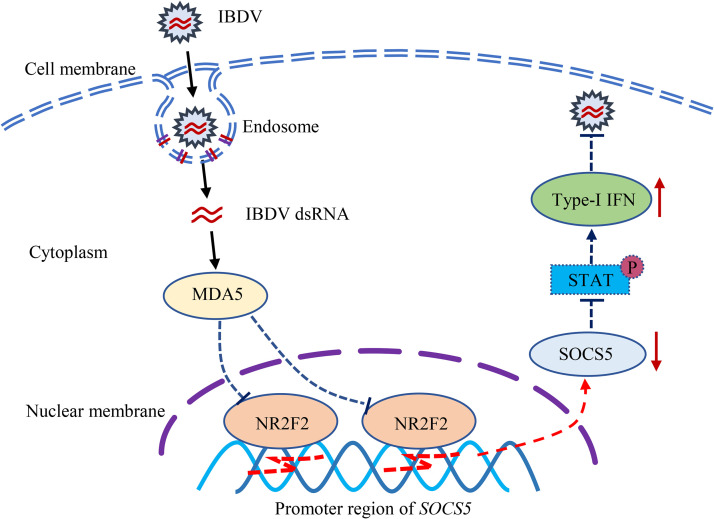
Nuclear receptors are ligand-activated transcription factors that play an important role in regulating innate antiviral immunity and other biological processes. However, the role of nuclear receptors in the host response to infectious bursal disease virus (IBDV) infection remains elusive. In this study, we show that IBDV infection or poly(I·C) treatment of DF-1 or HD11 cells markedly decreased nuclear receptor subfamily 2 group F member 2 (NR2F2) expression. Surprisingly, knockdown, knockout, or inhibition of NR2F2 expression in host cells remarkably inhibited IBDV replication and promoted IBDV/poly(I·C)-induced type I interferon and interferon-stimulated genes expression. Furthermore, our data show that NR2F2 negatively regulates the antiviral innate immune response by promoting the suppressor of cytokine signaling 5 (SOCS5) expression. Thus, reduced NR2F2 expression in the host response to IBDV infection inhibited viral replication by enhancing the expression of type I interferon by targeting SOCS5. These findings reveal that NR2F2 plays a crucial role in antiviral innate immunity, furthering our understanding of the mechanism underlying the host response to viral infection. IMPORTANCE Infectious bursal disease (IBD) is an immunosuppressive disease causing considerable economic losses to the poultry industry worldwide. Nuclear receptors play an important role in regulating innate antiviral immunity. However, the role of nuclear receptors in the host response to IBD virus (IBDV) infection remains elusive. Here, we report that NR2F2 expression decreased in IBDV-infected cells, which consequently reduced SOCS5 expression, promoted type I interferon expression, and suppressed IBDV infection. Thus, NR2F2 serves as a negative factor in the host response to IBDV infection by regulating SOCS5 expression, and intervention in the NR2F2-mediated host response by specific inhibitors might be employed as a strategy for prevention and treatment of IBD.
Keywords: IBDV; NR2F2; SOCS5; type I interferon.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
gga-miR-155 Enhances Type I Interferon Expression and Suppresses Infectious Burse Disease Virus Replication via Targeting SOCS1 and TANK.Front Cell Infect Microbiol. 2018 Mar 7;8:55. doi: 10.3389/fcimb.2018.00055. eCollection 2018. Front Cell Infect Microbiol. 2018. PMID: 29564226 Free PMC article.
-
gga-miR-9* inhibits IFN production in antiviral innate immunity by targeting interferon regulatory factor 2 to promote IBDV replication.Vet Microbiol. 2015 Jul 9;178(1-2):41-9. doi: 10.1016/j.vetmic.2015.04.023. Epub 2015 May 1. Vet Microbiol. 2015. PMID: 25975521
-
GATA3 Inhibits Viral Infection by Promoting MicroRNA-155 Expression.J Virol. 2022 Apr 13;96(7):e0188821. doi: 10.1128/jvi.01888-21. Epub 2022 Mar 23. J Virol. 2022. PMID: 35319228 Free PMC article.
-
Role of MicroRNAs in Host Defense against Infectious Bursal Disease Virus (IBDV) Infection: A Hidden Front Line.Viruses. 2020 May 14;12(5):543. doi: 10.3390/v12050543. Viruses. 2020. PMID: 32423052 Free PMC article. Review.
-
Host Combats IBDV Infection at Both Protein and RNA Levels.Viruses. 2022 Oct 21;14(10):2309. doi: 10.3390/v14102309. Viruses. 2022. PMID: 36298864 Free PMC article. Review.
Cited by
-
Anti-angiogenesis and anti-immunosuppression gene therapy through targeting COUP-TFII in an in situ glioblastoma mouse model.Cancer Gene Ther. 2024 Aug;31(8):1135-1150. doi: 10.1038/s41417-024-00799-z. Epub 2024 Jun 26. Cancer Gene Ther. 2024. PMID: 38926596
-
Chicken GSDME, a major pore-forming molecule responsible for RNA virus-induced pyroptosis in chicken.J Virol. 2025 Jan 31;99(1):e0158824. doi: 10.1128/jvi.01588-24. Epub 2024 Nov 22. J Virol. 2025. PMID: 39576037 Free PMC article.
References
-
- Fan L, Wu T, Wang Y, Hussain A, Jiang N, Gao L, Li K, Gao Y, Liu C, Cui H, Pan Q, Zhang Y, Wang X, Qi X. 2020. Novel variants of infectious bursal disease virus can severely damage the bursa of fabricius of immunized chickens. Vet Microbiol 240:108507. doi:10.1016/j.vetmic.2019.108507. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
