

Saturated fatty acid-enriched small extracellular vesicles mediate a crosstalk inducing liver inflammation and hepatocyte insulin resistance
- PMID: 37360906
- PMCID: PMC10285285
- DOI: 10.1016/j.jhepr.2023.100756
Saturated fatty acid-enriched small extracellular vesicles mediate a crosstalk inducing liver inflammation and hepatocyte insulin resistance
Abstract
Background & aims: Lipotoxicity triggers non-alcoholic fatty liver disease (NAFLD) progression owing to the accumulation of toxic lipids in hepatocytes including saturated fatty acids (SFAs), which activate pro-inflammatory pathways. We investigated the impact of hepatocyte- or circulating-derived small extracellular vesicles (sEV) secreted under NAFLD conditions on liver inflammation and hepatocyte insulin signalling.
Methods: sEV released by primary mouse hepatocytes, characterised and analysed by lipidomics, were added to mouse macrophages/Kupffer cells (KC) to monitor internalisation and inflammatory responses. Insulin signalling was analysed in hepatocytes exposed to conditioned media from sEV-loaded macrophages/KC. Mice were i.v. injected sEV to study liver inflammation and insulin signalling. Circulating sEV from mice and humans with NAFLD were used to evaluate macrophage-hepatocyte crosstalk.
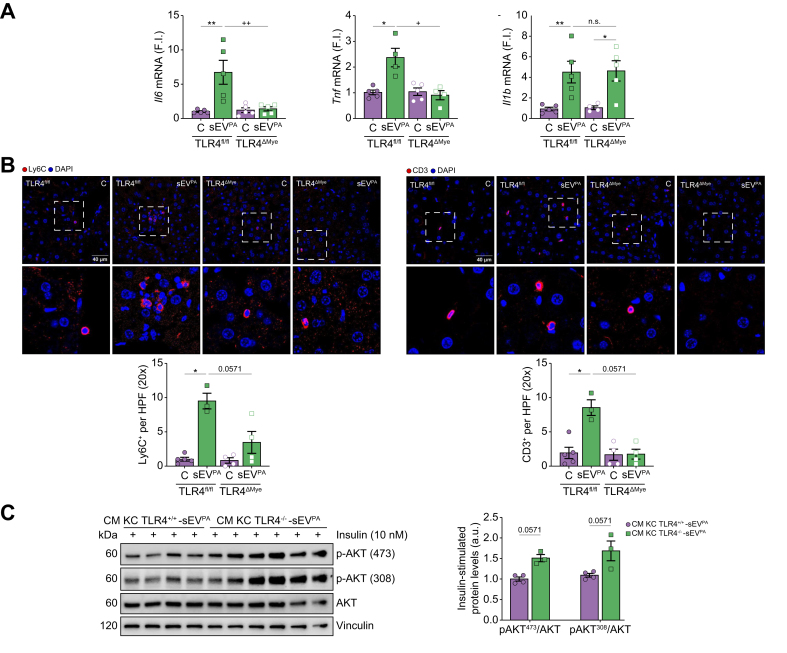
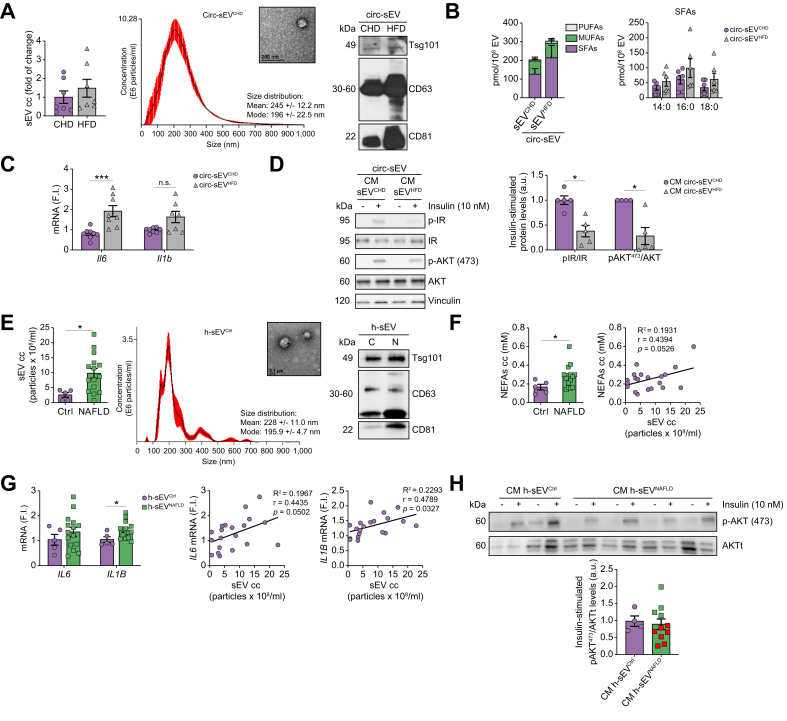
Results: Numbers of sEV released by hepatocytes increased under NAFLD conditions. Lipotoxic sEV were internalised by macrophages through the endosomal pathway and induced pro-inflammatory responses that were ameliorated by pharmacological inhibition or deletion of Toll-like receptor-4 (TLR4). Hepatocyte insulin signalling was impaired upon treatment with conditioned media from macrophages/KC loaded with lipotoxic sEV. Both hepatocyte-released lipotoxic sEV and the recipient macrophages/KC were enriched in palmitic (C16:0) and stearic (C18:0) SFAs, well-known TLR4 activators. Upon injection, lipotoxic sEV rapidly reached KC, triggering a pro-inflammatory response in the liver monitored by Jun N-terminal kinase (JNK) phosphorylation, NF-κB nuclear translocation, pro-inflammatory cytokine expression, and infiltration of immune cells into the liver parenchyma. sEV-mediated liver inflammation was attenuated by pharmacological inhibition or deletion of TLR4 in myeloid cells. Macrophage inflammation and subsequent hepatocyte insulin resistance were also induced by circulating sEV from mice and humans with NAFLD.
Conclusions: We identified hepatocyte-derived sEV as SFA transporters targeting macrophages/KC and activating a TLR4-mediated pro-inflammatory response enough to induce hepatocyte insulin resistance.
Impact and implications: Small extracellular vesicles (sEV) released by the hepatocytes under non-alcoholic fatty liver disease (NAFLD) conditions cause liver inflammation and insulin resistance in hepatocytes via paracrine hepatocyte-macrophage-hepatocyte crosstalk. We identified sEV as transporters of saturated fatty acids (SFAs) and potent lipotoxic inducers of liver inflammation. TLR4 deficiency or its pharmacological inhibition ameliorated liver inflammation induced by hepatocyte-derived lipotoxic sEV. Evidence of this macrophage-hepatocyte interactome was also found in patients with NAFLD, pointing to the relevance of sEV in SFA-mediated lipotoxicity in NAFLD.
Keywords: Extracellular vesicles; Inflammation; Insulin resistance; Lipotoxicity; NAFLD; TLR4.
© 2023 The Authors.
Conflict of interest statement
The authors declare no conflicts of interest related to this work. Please refer to the accompanying ICMJE disclosure forms for further details.
Figures







References
-
- Younossi Z.M., Koenig A.B., Abdelatif D., Fazel Y., Henry L., Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. - PubMed
-
- Eslam M., Newsome P.N., Sarin S.K., Anstee Q.M., Targher G., Romero-Gomez M., et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73:202–209. - PubMed
-
- Adams L.A., Ratziu V. Non-alcoholic fatty liver – perhaps not so benign. J Hepatol. 2015;62:1002–1004. - PubMed
-
- Béland-Bonenfant S., Rouland A., Petit J.M., Vergès B. Concise review of lipidomics in nonalcoholic fatty liver disease. Diabetes Metab. 2023;49 - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
