

A novel dual NLRP1 and NLRP3 inflammasome inhibitor for the treatment of inflammatory diseases
- PMID: 37360982
- PMCID: PMC10288073
- DOI: 10.1002/cti2.1455
A novel dual NLRP1 and NLRP3 inflammasome inhibitor for the treatment of inflammatory diseases
Abstract
Objectives: Inflammasomes induce maturation of the inflammatory cytokines IL-1β and IL-18, whose activity is associated with the pathophysiology of a wide range of infectious and inflammatory diseases. As validated therapeutic targets for the treatment of acute and chronic inflammatory diseases, there has been intense interest in developing small-molecule inhibitors to target inflammasome activity and reduce disease-associated inflammatory burden.
Methods: We examined the therapeutic potential of a novel small-molecule inhibitor, and associated derivatives, termed ADS032 to target and reduce inflammasome-mediated inflammation in vivo. In vitro, we characterised ADS032 function, target engagement and specificity.
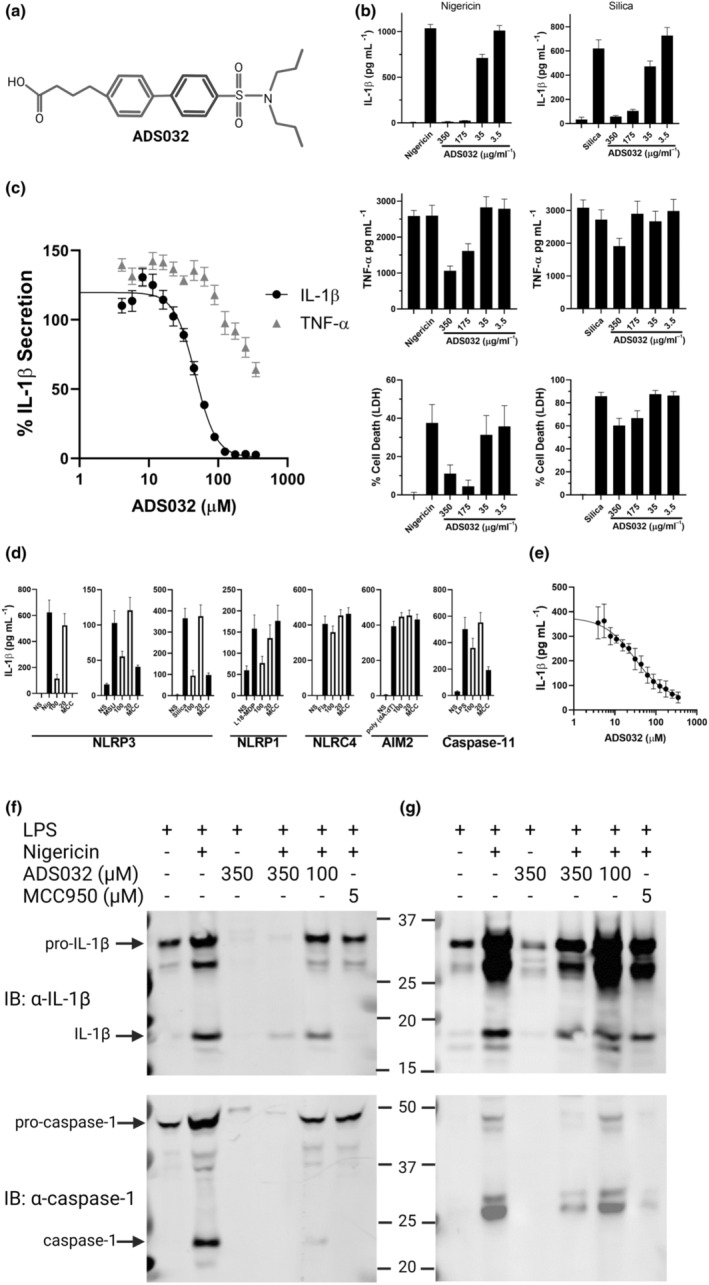
Results: We describe ADS032 as the first dual NLRP1 and NLRP3 inhibitor. ADS032 is a rapid, reversible and stable inflammasome inhibitor that directly binds both NLRP1 and NLRP3, reducing secretion and maturation of IL-1β in human-derived macrophages and bronchial epithelial cells in response to the activation of NLPR1 and NLRP3. ADS032 also reduced NLRP3-induced ASC speck formation, indicative of targeting inflammasome formation. In vivo, ADS032 reduced IL-1β and TNF-α levels in the serum of mice challenged i.p. with LPS and reduced pulmonary inflammation in an acute model of lung silicosis. Critically, ADS032 protected mice from lethal influenza A virus challenge, displayed increased survival and reduced pulmonary inflammation.
Conclusion: ADS032 is the first described dual inflammasome inhibitor and a potential therapeutic to treat both NLRP1- and NLRP3-associated inflammatory diseases and also constitutes a novel tool that allows examination of the role of NLRP1 in human disease.
Keywords: NLRP1; NLRP3; drug targets; inflammasome; inflammation; pulmonary inflammation.
© 2023 Adiso Therapeutics and The Authors. Clinical & Translational Immunology published by John Wiley & Sons Australia, Ltd on behalf of Australian and New Zealand Society for Immunology, Inc.
Conflict of interest statement
RED and MAN receive consultancy fees from Adiso Therapeutics Inc. RF, DLF, CKM and AM are employees of Adiso Therapeutics Inc. AJF and AGR are supported by an Adiso Therapeutics Inc research grant to the University of Edinburgh Centre for Inflammation Research. RED, MAN, RF and CKM have patent ownership, while CKM and AM retain stock ownership in Adiso Therapeutics.
Figures







References
-
- Pandey A, Shen C, Feng S, Man SM. Cell biology of inflammasome activation. Trends Cell Biol 2021; 31: 924–939. - PubMed
-
- Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140: 821–832. - PubMed
-
- Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016; 16: 407–420. - PubMed
-
- Ross C, Chan AH, von Pein JB, Maddugoda MP, Boucher D, Schroder K. Inflammatory caspases: toward a unified model for caspase activation by inflammasomes. Annu Rev Immunol 2022; 40: 249–269. - PubMed
-
- Broz P, Pelegrin P, Shao F. The gasdermin, a protein family executing cell death and inflammation. Nat Rev Immunol 2020; 20: 143–157. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous