

A case of hyperphosphatemic familial tumoral calcinosis due to maternal uniparental disomy of a GALNT3 variant
- PMID: 37362161
- PMCID: PMC10288290
- DOI: 10.1297/cpe.2022-0071
A case of hyperphosphatemic familial tumoral calcinosis due to maternal uniparental disomy of a GALNT3 variant
Abstract
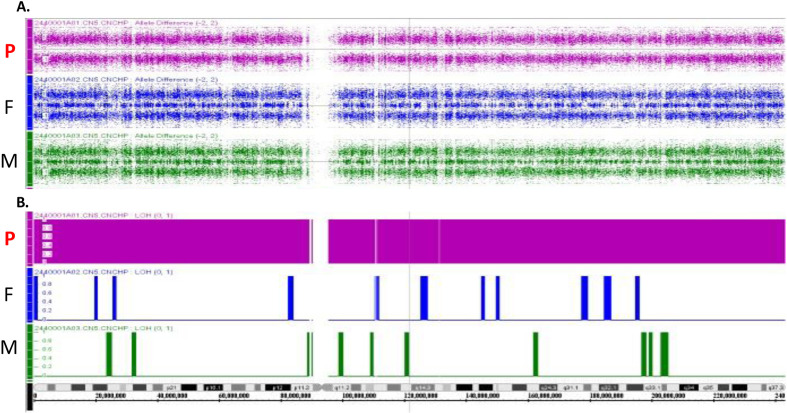
Hyperphosphatemic familial tumoral calcinosis (HFTC) is a rare, inherited autosomal recessive disorder caused by fibroblast growth factor-23 (FGF23), N-acetylgalactosaminyltransferase 3 (GALNT3), or Klotho (KL) gene variants. Here, we report the case of a Japanese boy who presented with a mass in his left elbow at the age of three. Laboratory test results of the patient revealed normocalcemia (10.3 mg/dL) and hyperphosphatemia (8.7 mg/dL); however, despite hyperphosphatemia, serum intact FGF23 level was low, renal tubular reabsorption of phosphate (TRP) level was inappropriately increased, and 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) level was inappropriately normal. Genetic analysis revealed maternal uniparental disomy (UPD) of chromosome 2, which included a novel GALNT3 variant (c.1780-1G>C). Reverse transcription-polymerase chain reaction (RT-PCR) analysis of GALNT3 mRNA confirmed that this variant resulted in the destruction of exon 11. We resected the mass when the patient was five years old, owing to its gradual enlargement. No relapse or new pathological lesions were observed four years after tumor resection. This is the first case report of a Japanese patient with HFTC associated with a novel GALNT3 variant, as well as the first case of HFTC caused by maternal UPD of chromosome 2 that includes the GALNT3 variant.
Keywords: N-acetylgalactosaminyltransferase 3 (GALNT3); hyperostosis–hyperphosphatemia syndrome; hyperphosphatemic familial tumoral calcinosis; tumoral calcinosis; uniparental disomy.
2023©The Japanese Society for Pediatric Endocrinology.
Figures


References
Publication types
LinkOut - more resources
Full Text Sources