

5-Aminolevulinic acid bypasses mitochondrial complex I deficiency and corrects physiological dysfunctions in Drosophila
- PMID: 37364055
- PMCID: PMC10407699
- DOI: 10.1093/hmg/ddad092
5-Aminolevulinic acid bypasses mitochondrial complex I deficiency and corrects physiological dysfunctions in Drosophila
Abstract
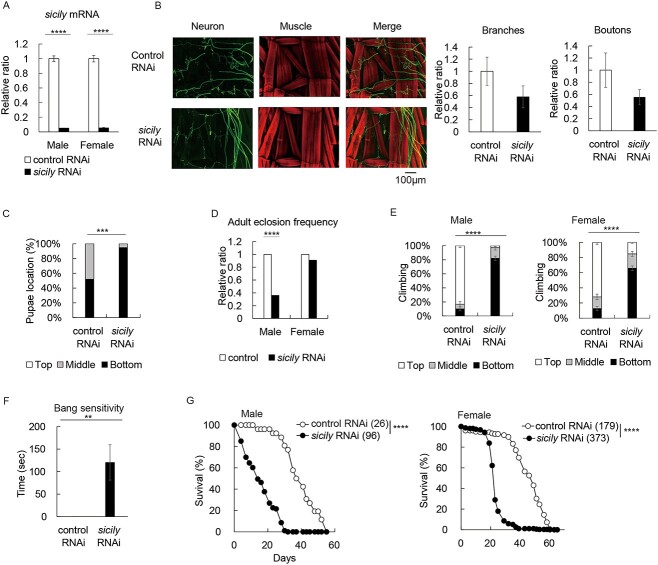
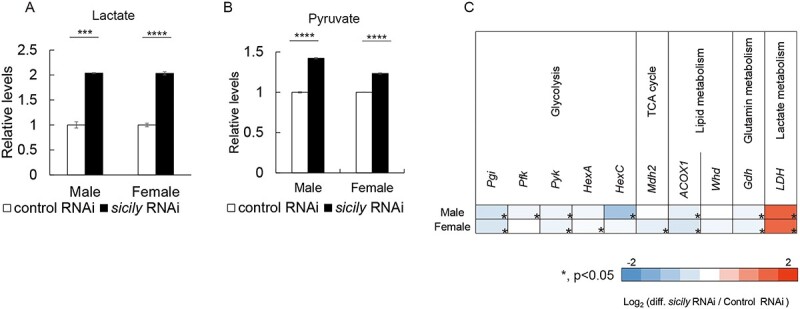
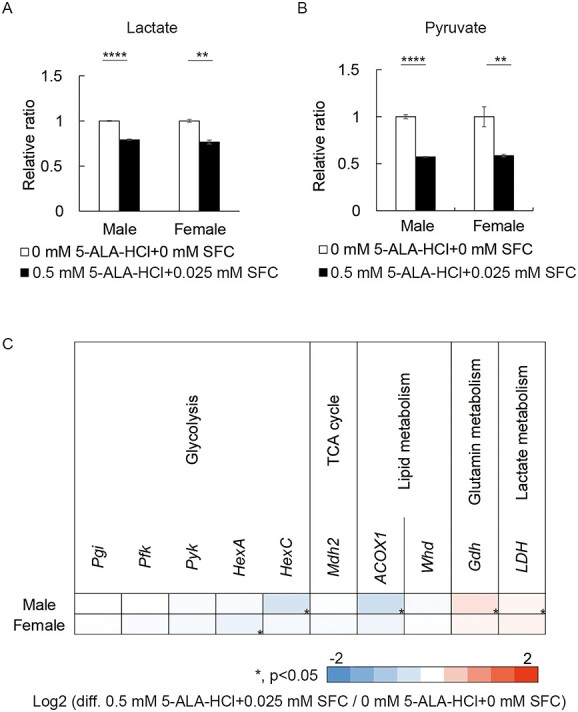
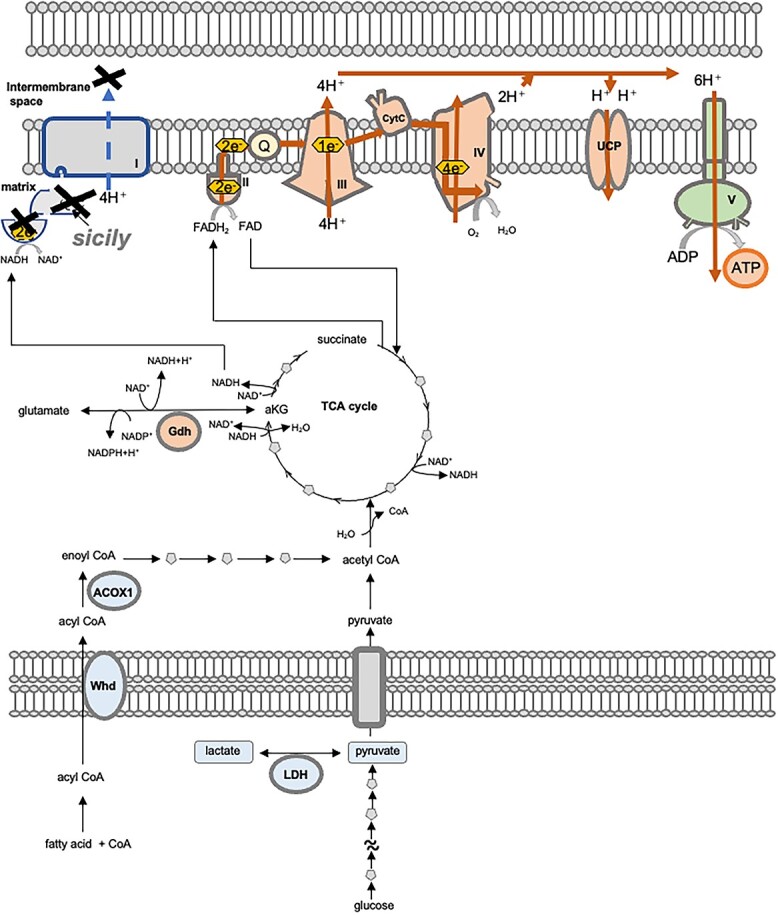
Complex I (CI) deficiency in mitochondrial oxidative phosphorylation (OXPHOS) is the most common cause of mitochondrial diseases, and limited evidence-based treatment options exist. Although CI provides the most electrons to OXPHOS, complex II (CII) is another entry point of electrons. Enhancement of this pathway may compensate for a loss of CI; however, the effects of boosting CII activity on CI deficiency are unclear at the animal level. 5-Aminolevulinic acid (5-ALA) is a crucial precursor of heme, which is essential for CII, complex III, complex IV (CIV) and cytochrome c activities. Here, we show that feeding a combination of 5-ALA hydrochloride and sodium ferrous citrate (5-ALA-HCl + SFC) increases ATP production and suppresses defective phenotypes in Drosophila with CI deficiency. Knockdown of sicily, a Drosophila homolog of the critical CI assembly protein NDUFAF6, caused CI deficiency, accumulation of lactate and pyruvate and detrimental phenotypes such as abnormal neuromuscular junction development, locomotor dysfunctions and premature death. 5-ALA-HCl + SFC feeding increased ATP levels without recovery of CI activity. The activities of CII and CIV were upregulated, and accumulation of lactate and pyruvate was suppressed. 5-ALA-HCl + SFC feeding improved neuromuscular junction development and locomotor functions in sicily-knockdown flies. These results suggest that 5-ALA-HCl + SFC shifts metabolic programs to cope with CI deficiency. Bullet outline 5-Aminolevulinic acid (5-ALA-HCl + SFC) increases ATP production in flies with complex I deficiency.5-ALA-HCl + SFC increases the activities of complexes II and IV.5-ALA-HCl + SFC corrects metabolic abnormalities and suppresses the detrimental phenotypes caused by complex I deficiency.
© The Author(s) 2023. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures



References
-
- Murayama, K., Shimura, M., Liu, Z., Okazaki, Y. and Ohtake, A. (2019) Recent topics: the diagnosis, molecular genesis, and treatment of mitochondrial diseases. J. Hum. Genet., 64, 113–125. - PubMed
-
- Gorman, G.S., Chinnery, P.F., DiMauro, S., Hirano, M., Koga, Y., McFarland, R., Suomalainen, A., Thorburn, D.R., Zeviani, M. and Turnbull, D.M. (2016) Mitochondrial diseases. Nat. Rev. Dis. Primers, 2, 16080. - PubMed
-
- Cogliati, S., Enriquez, J.A. and Scorrano, L. (2016) Mitochondrial cristae: where beauty meets functionality. Trends Biochem. Sci., 41, 261–273. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
