Multi-level analysis of the gut-brain axis shows autism spectrum disorder-associated molecular and microbial profiles
- PMID: 37365313
- PMCID: PMC10322709
- DOI: 10.1038/s41593-023-01361-0
Multi-level analysis of the gut-brain axis shows autism spectrum disorder-associated molecular and microbial profiles
Abstract
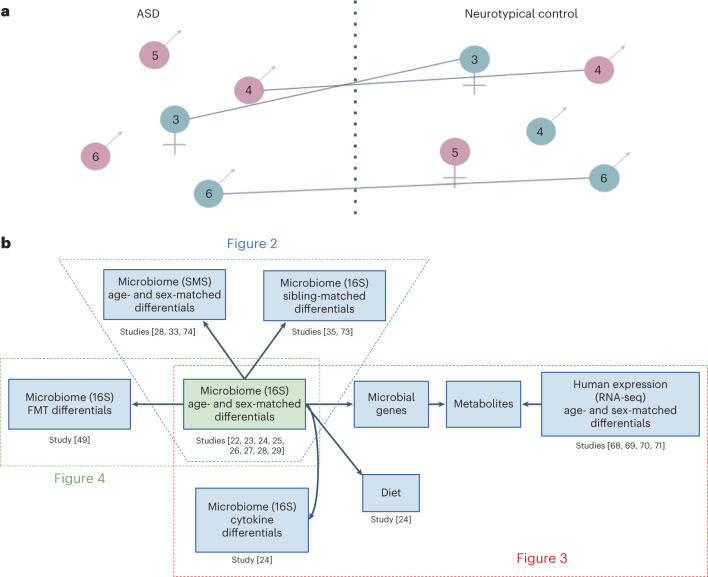
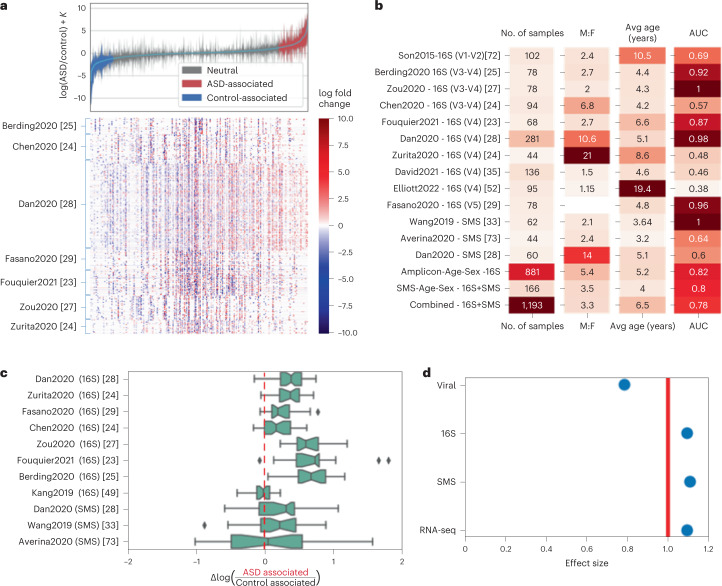
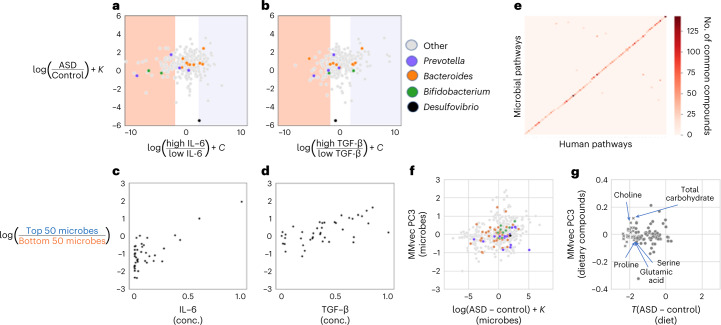
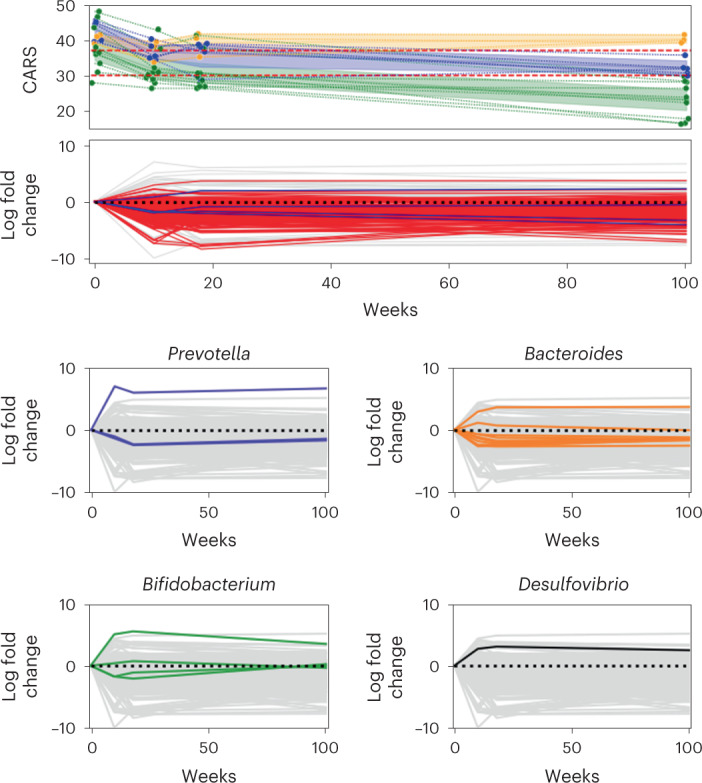
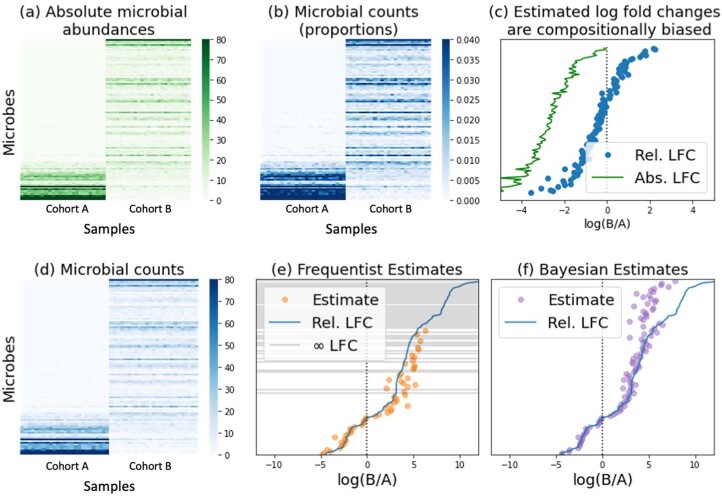
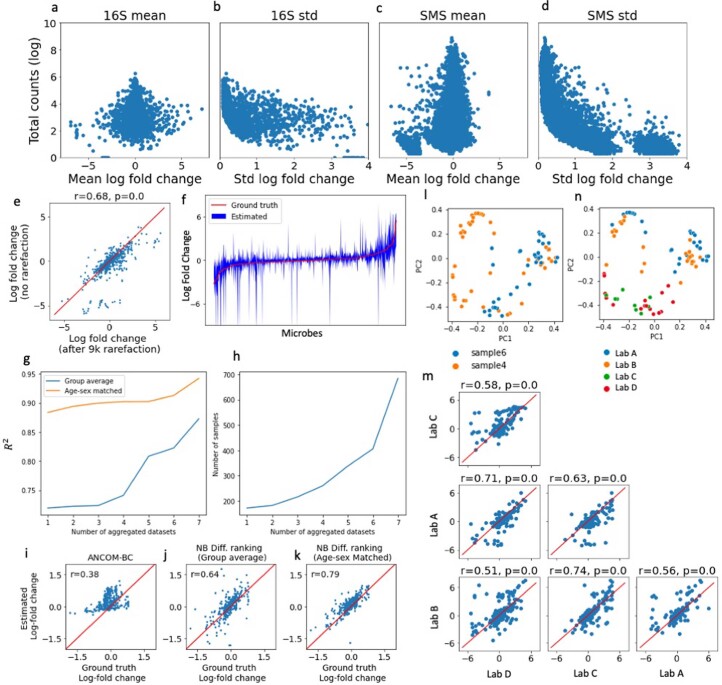
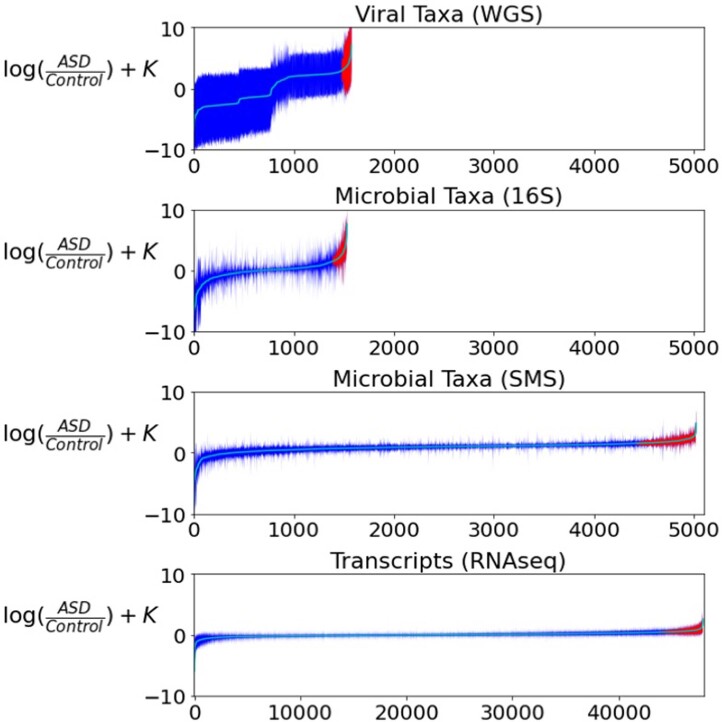
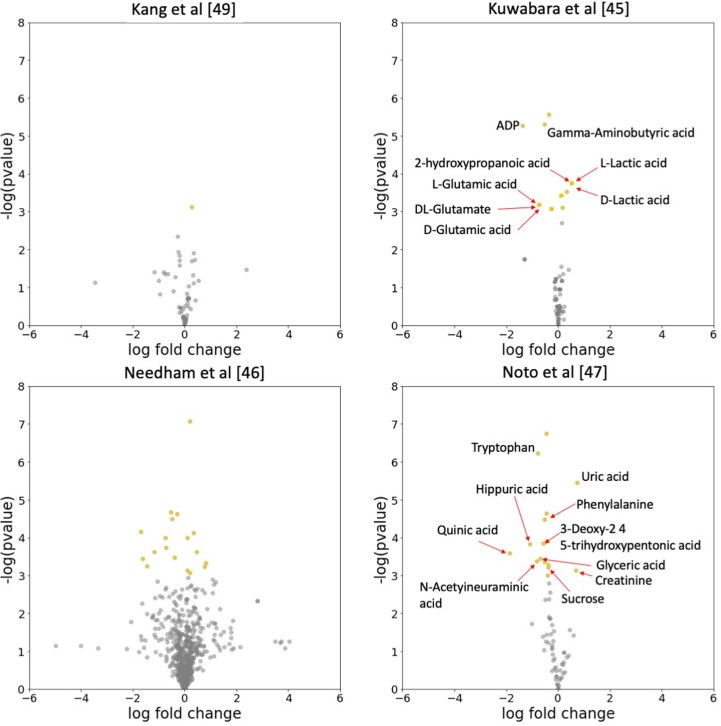
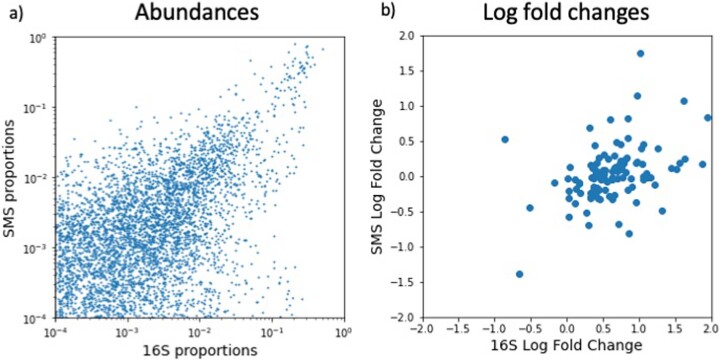
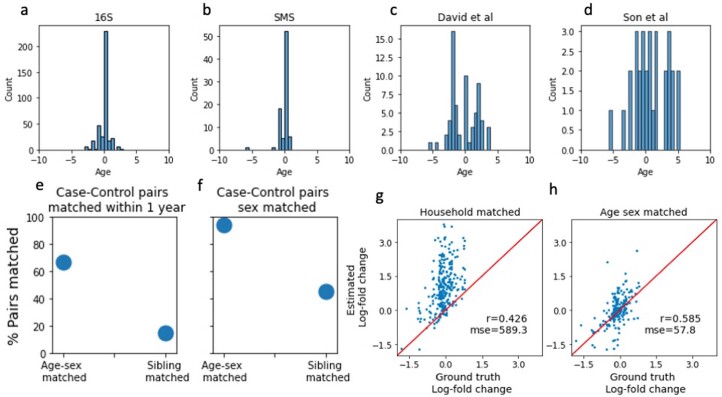
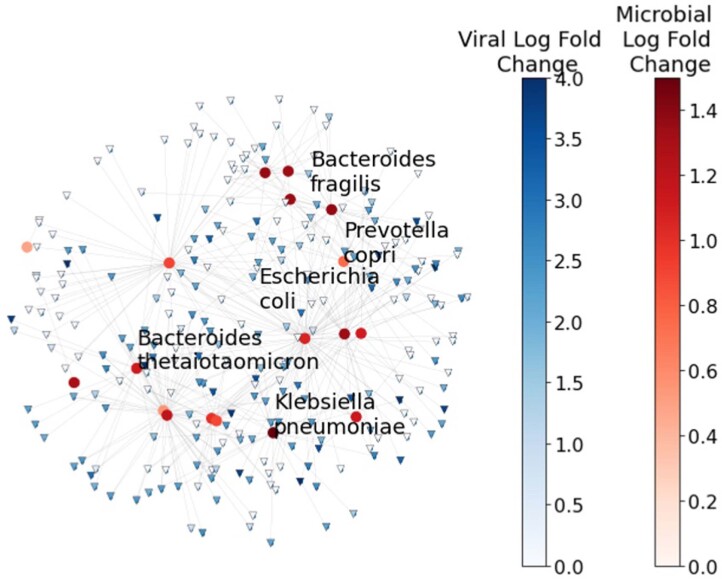
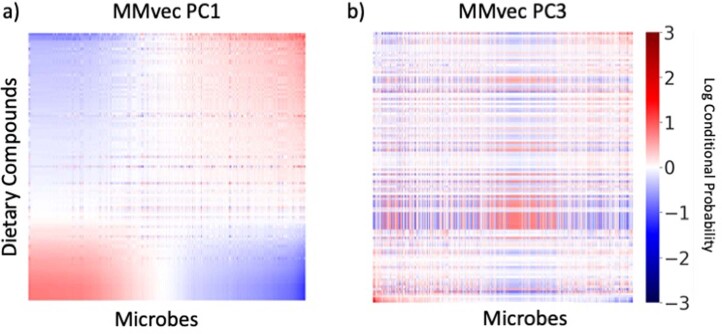
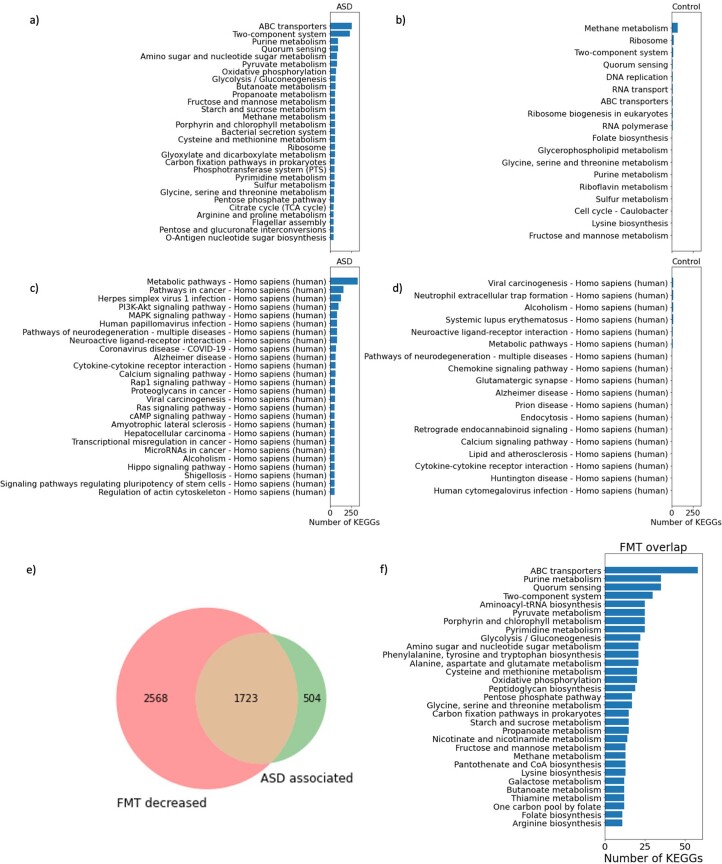
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by heterogeneous cognitive, behavioral and communication impairments. Disruption of the gut-brain axis (GBA) has been implicated in ASD although with limited reproducibility across studies. In this study, we developed a Bayesian differential ranking algorithm to identify ASD-associated molecular and taxa profiles across 10 cross-sectional microbiome datasets and 15 other datasets, including dietary patterns, metabolomics, cytokine profiles and human brain gene expression profiles. We found a functional architecture along the GBA that correlates with heterogeneity of ASD phenotypes, and it is characterized by ASD-associated amino acid, carbohydrate and lipid profiles predominantly encoded by microbial species in the genera Prevotella, Bifidobacterium, Desulfovibrio and Bacteroides and correlates with brain gene expression changes, restrictive dietary patterns and pro-inflammatory cytokine profiles. The functional architecture revealed in age-matched and sex-matched cohorts is not present in sibling-matched cohorts. We also show a strong association between temporal changes in microbiome composition and ASD phenotypes. In summary, we propose a framework to leverage multi-omic datasets from well-defined cohorts and investigate how the GBA influences ASD.
© 2023. The Author(s).
Conflict of interest statement
R.H.M. is Scientific Director at Precidiag, Inc. T.D.L. is a co-founder and Chief Scientific Officer of Microbiotica. S.K.M. is a co-founder and has equity in Axial Therapeutics. R.J.X. is a co-founder of Celsius Therapeutics and Jnana Therapeutics, a member of the Scientific Advisory Board at Nestle and a member of the Board of Directors at Moonlake Immunotherapeutics. R.B. is currently Executive Director of Prescient Design, a Genentech Accelerator. J.T.M. is the founder of Gutz Analytics and a co-founder of Integrated Omics AI. G.T.-O. is a Consultant-in-Residence at the Simons Foundation. The remaining authors declare no competing interests.
Figures