

Transcriptome- and proteome-wide association studies nominate determinants of kidney function and damage
- PMID: 37365616
- PMCID: PMC10291807
- DOI: 10.1186/s13059-023-02993-y
Transcriptome- and proteome-wide association studies nominate determinants of kidney function and damage
Abstract
Background: The pathophysiological causes of kidney disease are not fully understood. Here we show that the integration of genome-wide genetic, transcriptomic, and proteomic association studies can nominate causal determinants of kidney function and damage.
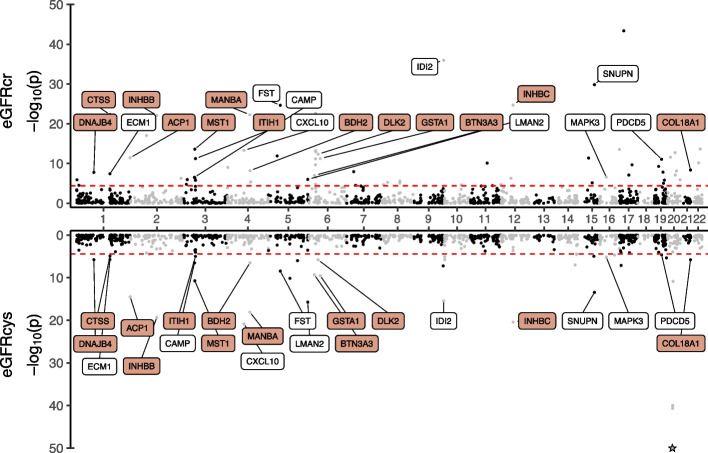
Results: Through transcriptome-wide association studies (TWAS) in kidney cortex, kidney tubule, liver, and whole blood and proteome-wide association studies (PWAS) in plasma, we assess for effects of 12,893 genes and 1342 proteins on kidney filtration (glomerular filtration rate (GFR) estimated by creatinine; GFR estimated by cystatin C; and blood urea nitrogen) and kidney damage (albuminuria). We find 1561 associations distributed among 260 genomic regions that are supported as putatively causal. We then prioritize 153 of these genomic regions using additional colocalization analyses. Our genome-wide findings are supported by existing knowledge (animal models for MANBA, DACH1, SH3YL1, INHBB), exceed the underlying GWAS signals (28 region-trait combinations without significant GWAS hit), identify independent gene/protein-trait associations within the same genomic region (INHBC, SPRYD4), nominate tissues underlying the associations (tubule expression of NRBP1), and distinguish markers of kidney filtration from those with a role in creatinine and cystatin C metabolism. Furthermore, we follow up on members of the TGF-beta superfamily of proteins and find a prognostic value of INHBC for kidney disease progression even after adjustment for measured glomerular filtration rate (GFR).
Conclusion: In summary, this study combines multimodal, genome-wide association studies to generate a catalog of putatively causal target genes and proteins relevant to kidney function and damage which can guide follow-up studies in physiology, basic science, and clinical medicine.
Keywords: ACR; BUN; Chronic kidney disease; End-stage kidney disease; GWAS; Genetics; Nephrology; PWAS; Proteomics; TWAS; Transcriptomics; eGFR.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
-
- Stevens, P.E., Levin, A. & Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group, M. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann Intern Med. 2013; 158: 825–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
