

Hypomorphic pathogenic variant in SFTPB leads to adult pulmonary fibrosis
- PMID: 37380697
- PMCID: PMC10474257
- DOI: 10.1038/s41431-023-01413-w
Hypomorphic pathogenic variant in SFTPB leads to adult pulmonary fibrosis
Abstract
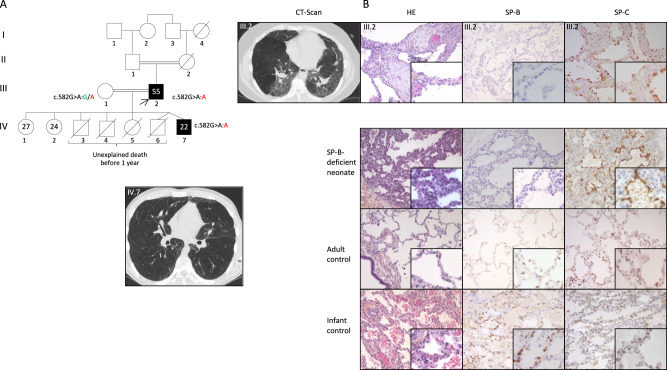
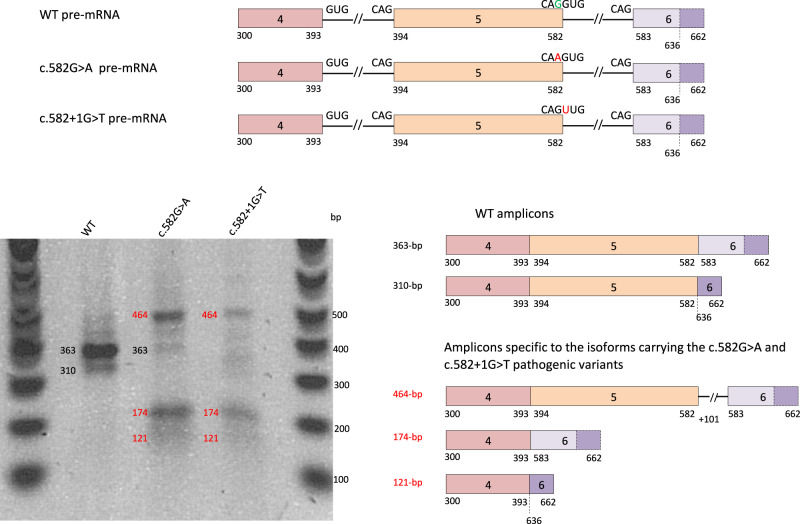
Biallelic pathogenic variants in the surfactant protein (SP)-B gene (SFTPB) have been associated with fatal forms of interstitial lung diseases (ILD) in newborns and exceptional survival in young children. We herein report the cases of two related adults with pulmonary fibrosis due to a new homozygous SFTPB pathogenic variant, c.582G>A p.(Gln194=). In vitro transcript studies showed that this SFTPB synonymous pathogenic variant induces aberrant splicing leading to three abnormal transcripts with the preservation of the expression of a small proportion of normal SFTPB transcripts. Immunostainings on lung biopsies of the proband showed an almost complete loss of SP-B expression. This hypomorphic splice variant has thus probably allowed the patients' survival to adulthood while inducing an epithelial cell dysfunction leading to ILD. Altogether, this report shows that SFTPB pathogenic variants should be considered in atypical presentations and/or early-onset forms of ILD particularly when a family history is identified.
© 2023. The Author(s), under exclusive licence to European Society of Human Genetics.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
