

Locus for severity implicates CNS resilience in progression of multiple sclerosis
- PMID: 37380766
- PMCID: PMC10602210
- DOI: 10.1038/s41586-023-06250-x
Locus for severity implicates CNS resilience in progression of multiple sclerosis
Abstract
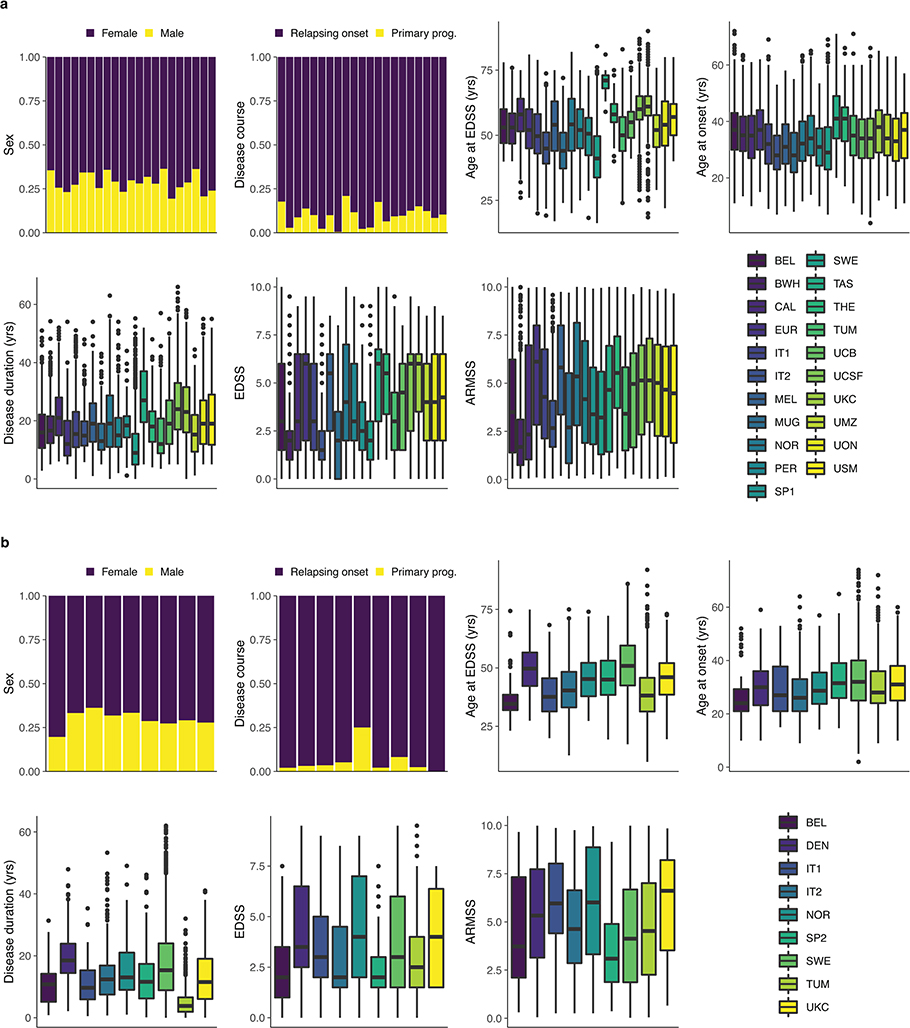
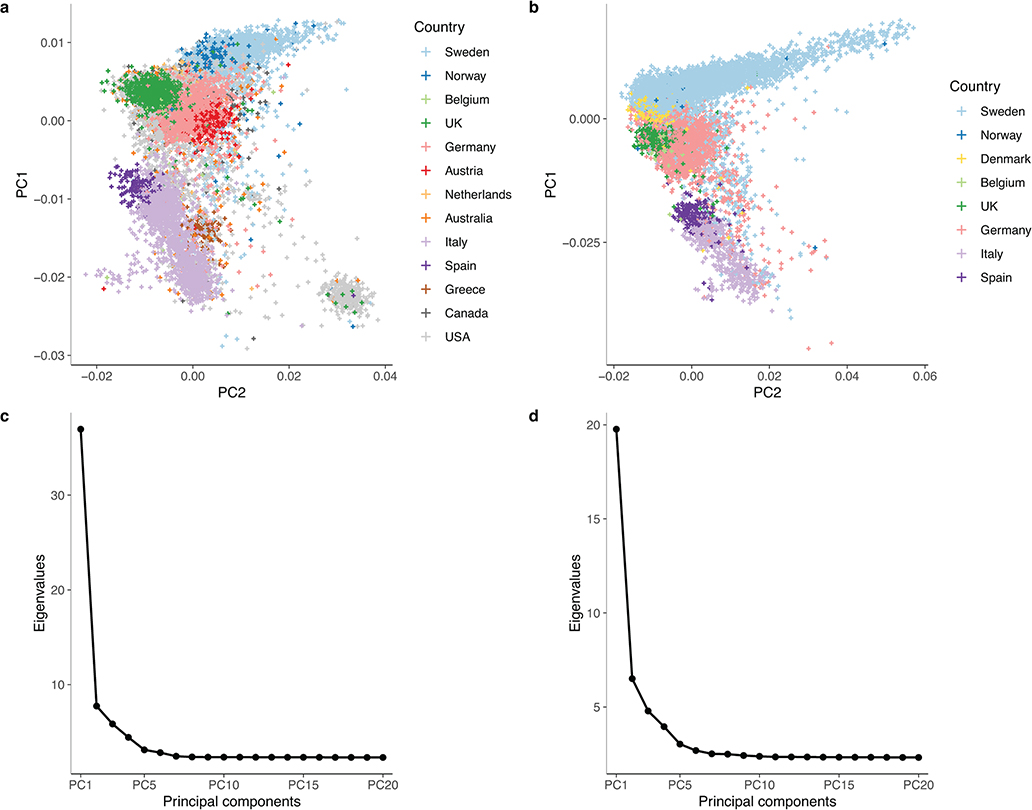
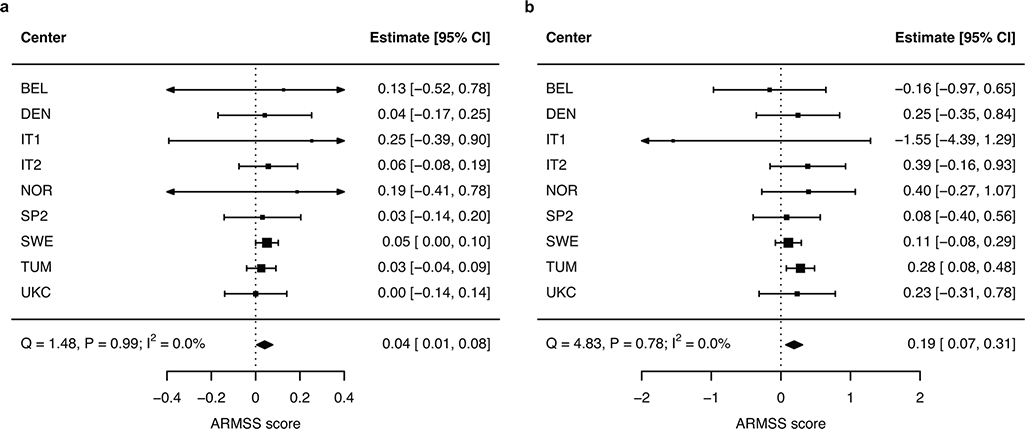
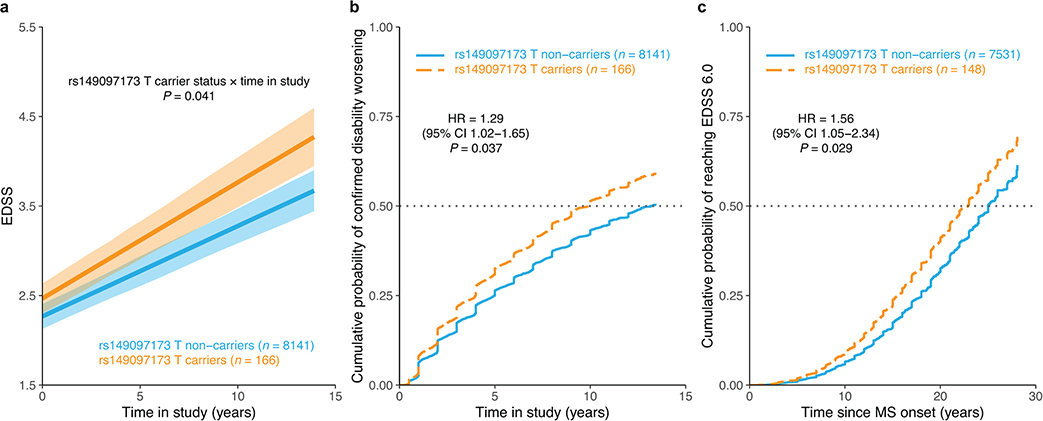
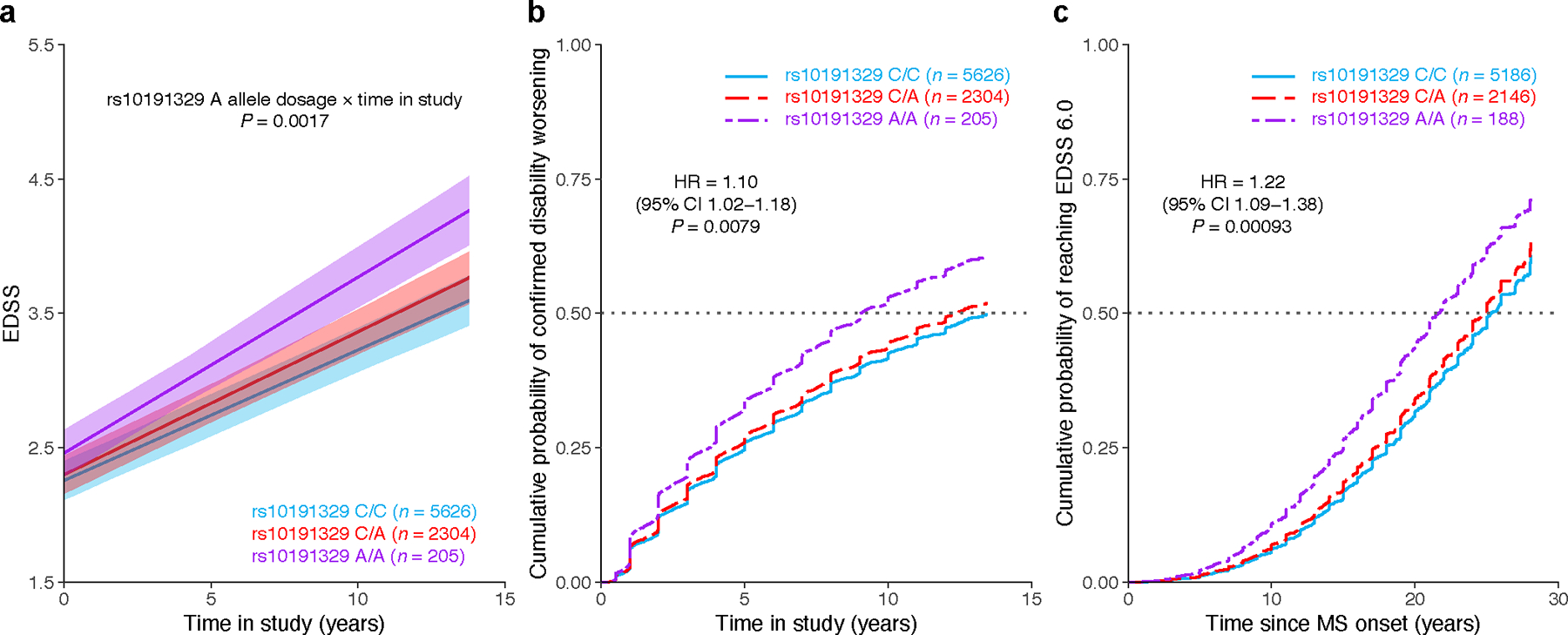
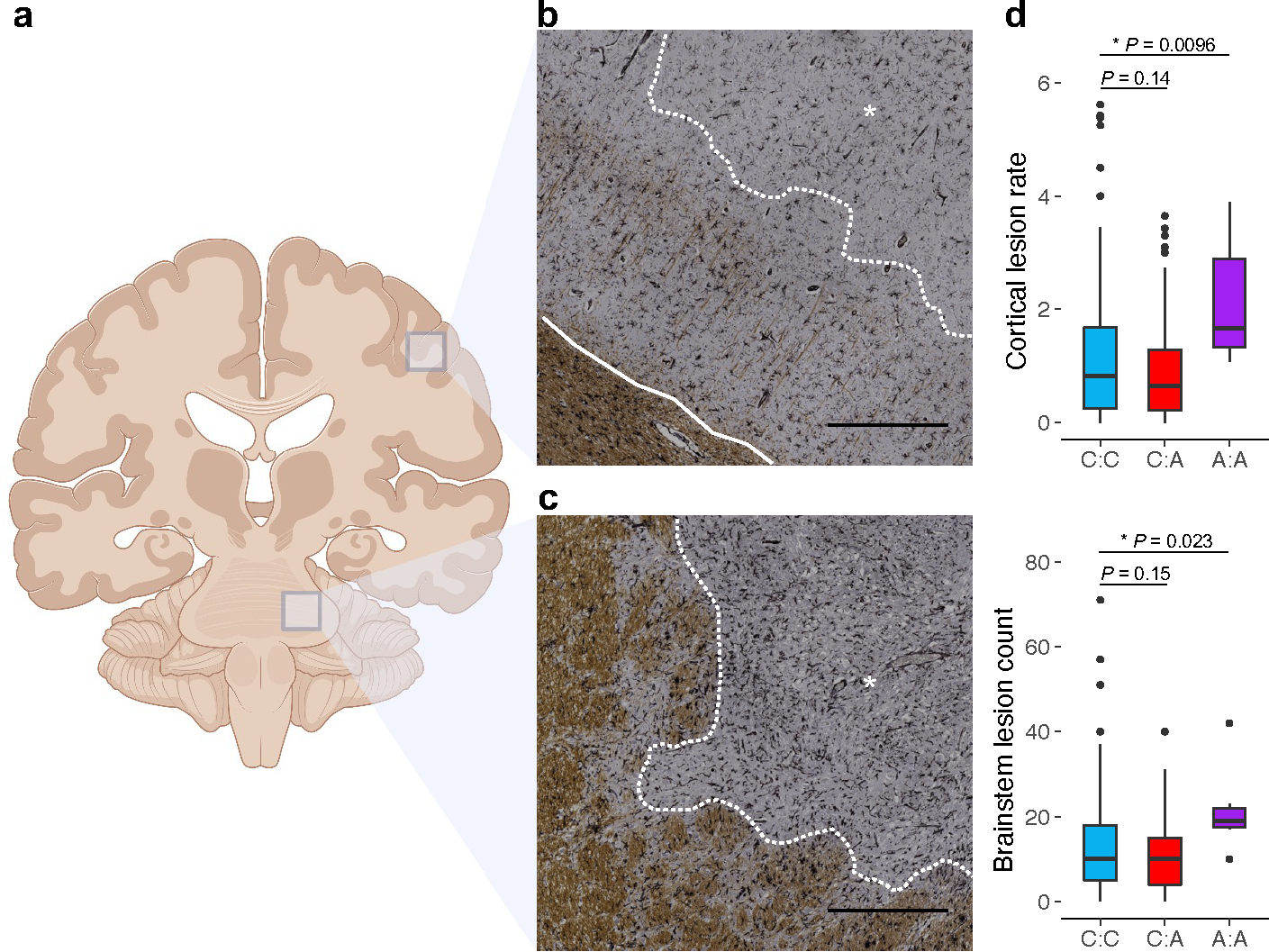
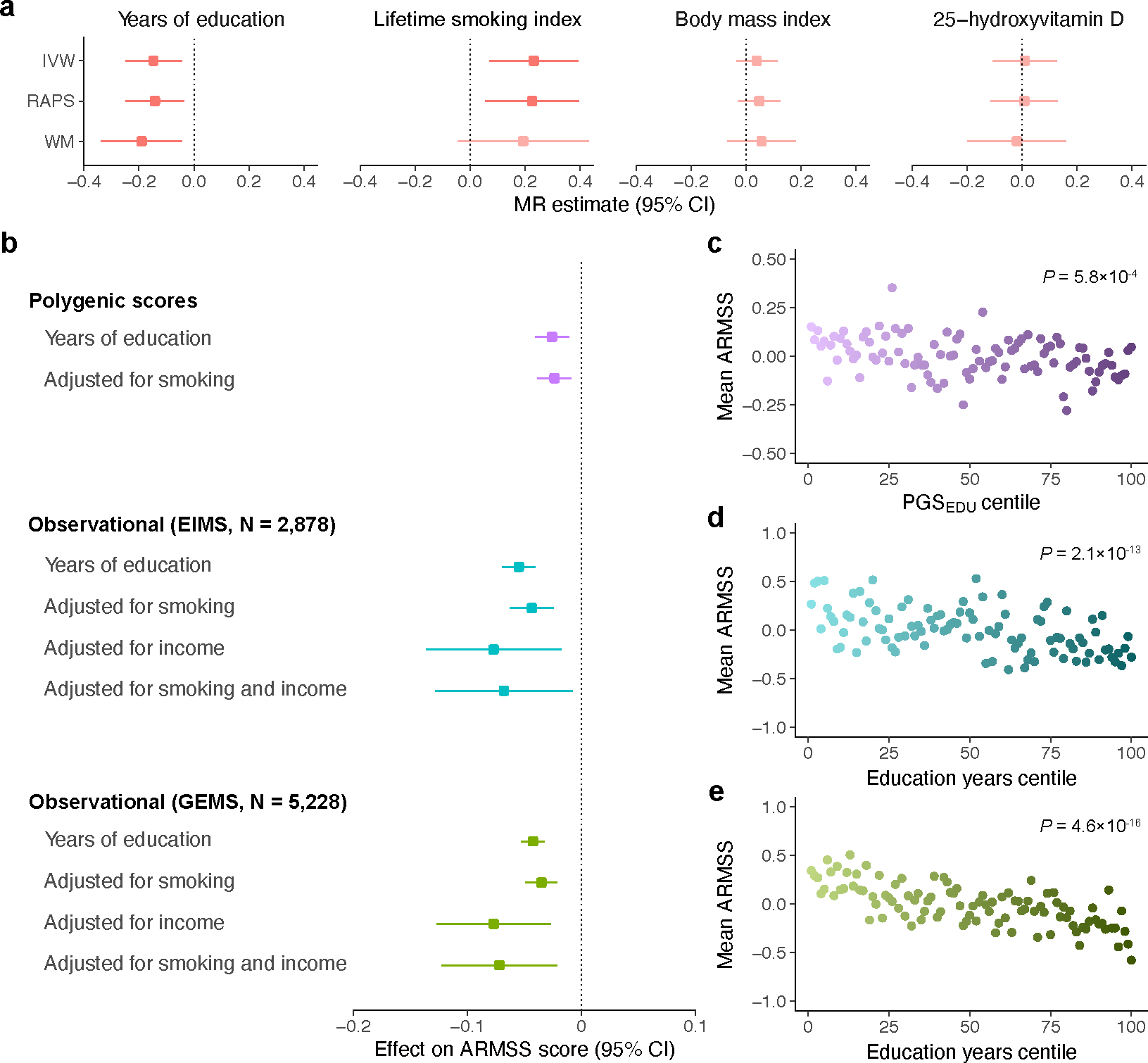
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system (CNS) that results in significant neurodegeneration in the majority of those affected and is a common cause of chronic neurological disability in young adults1,2. Here, to provide insight into the potential mechanisms involved in progression, we conducted a genome-wide association study of the age-related MS severity score in 12,584 cases and replicated our findings in a further 9,805 cases. We identified a significant association with rs10191329 in the DYSF-ZNF638 locus, the risk allele of which is associated with a shortening in the median time to requiring a walking aid of a median of 3.7 years in homozygous carriers and with increased brainstem and cortical pathology in brain tissue. We also identified suggestive association with rs149097173 in the DNM3-PIGC locus and significant heritability enrichment in CNS tissues. Mendelian randomization analyses suggested a potential protective role for higher educational attainment. In contrast to immune-driven susceptibility3, these findings suggest a key role for CNS resilience and potentially neurocognitive reserve in determining outcome in MS.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures
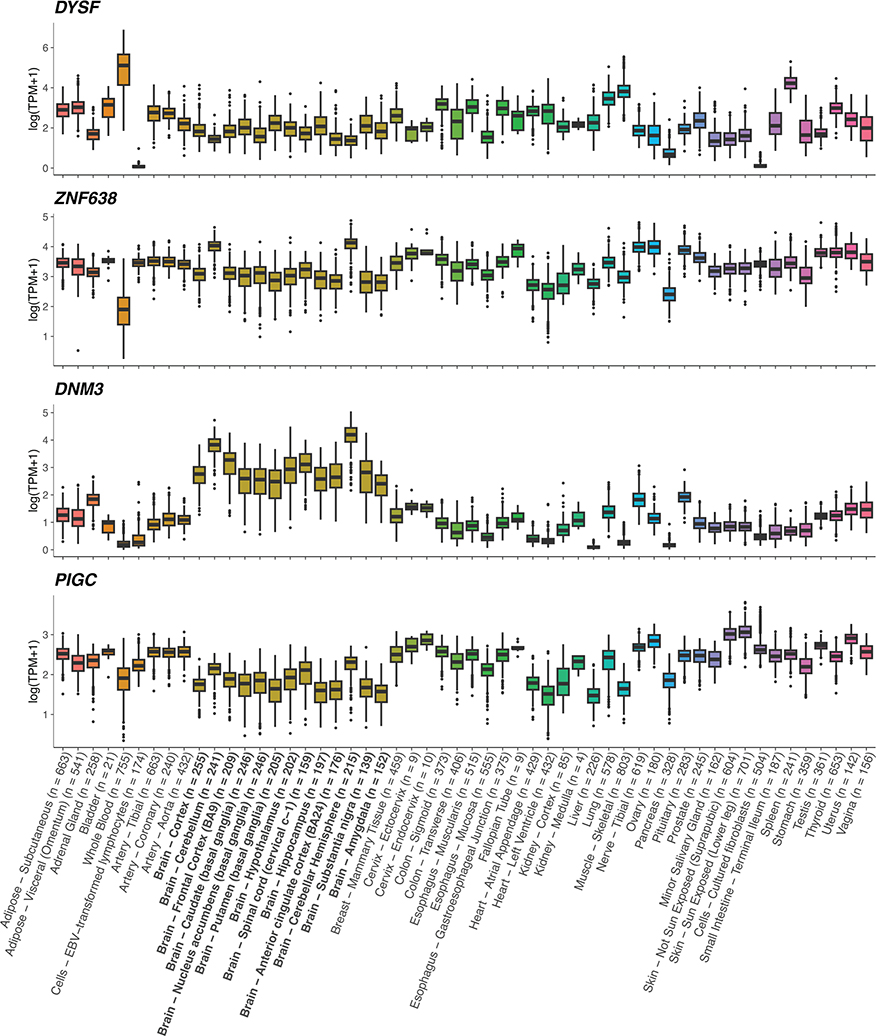
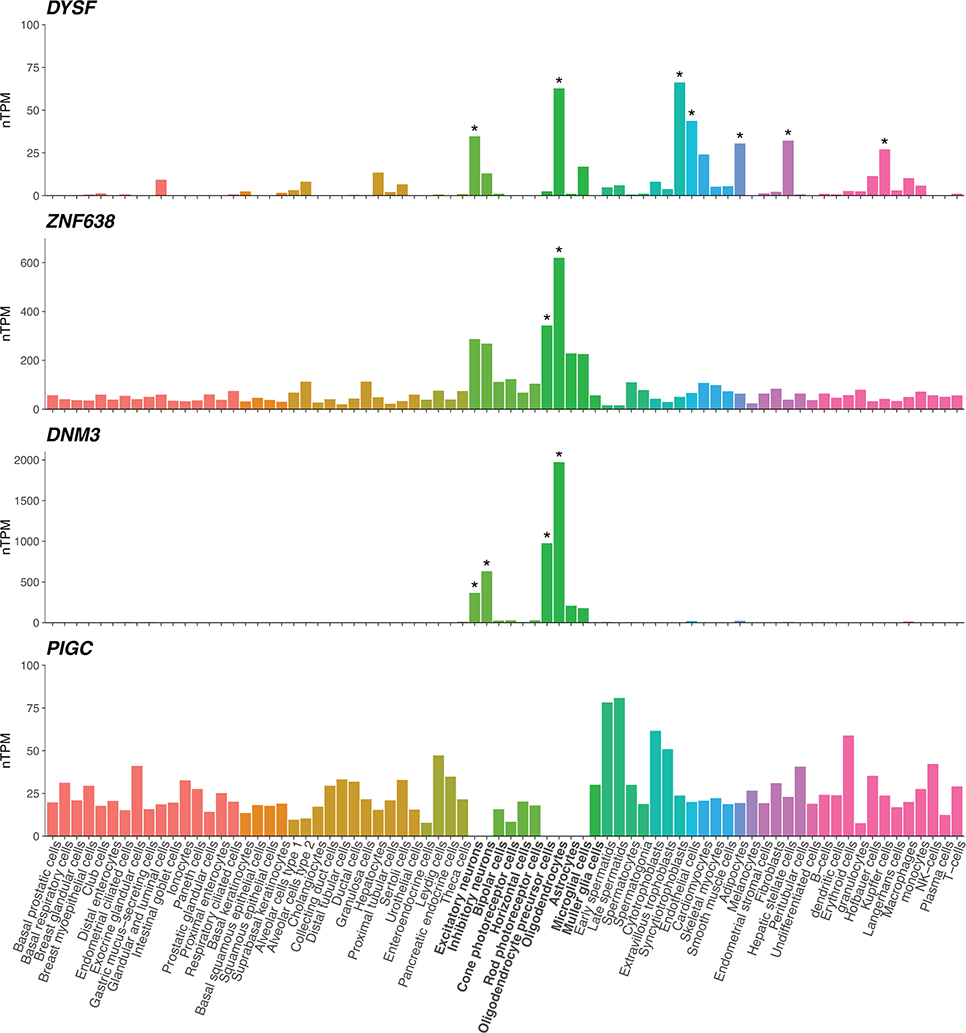
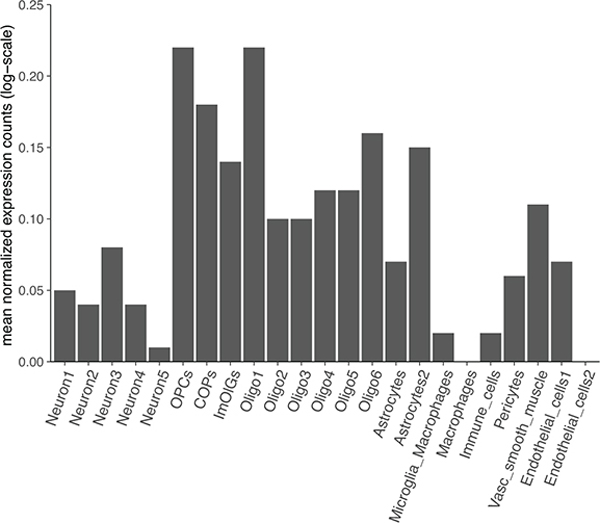
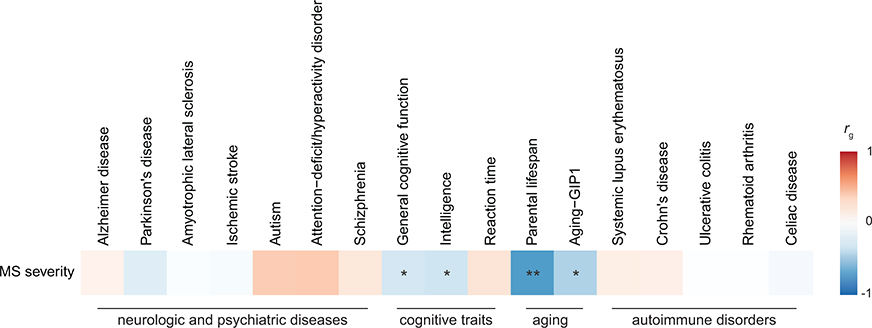
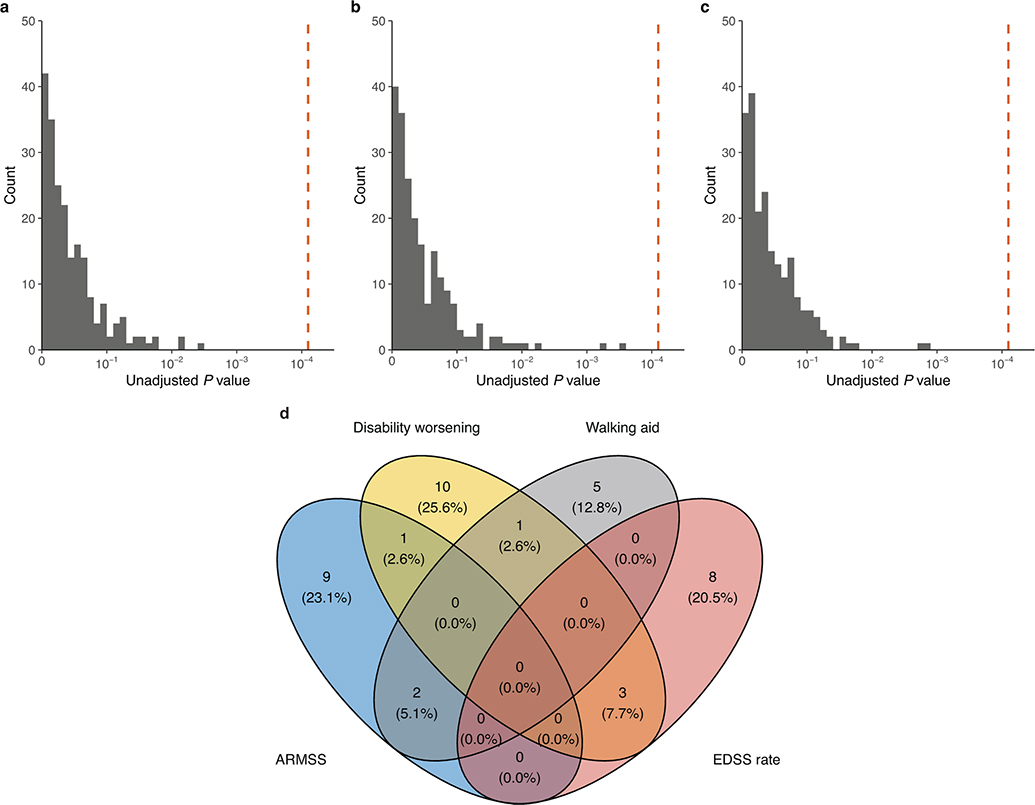
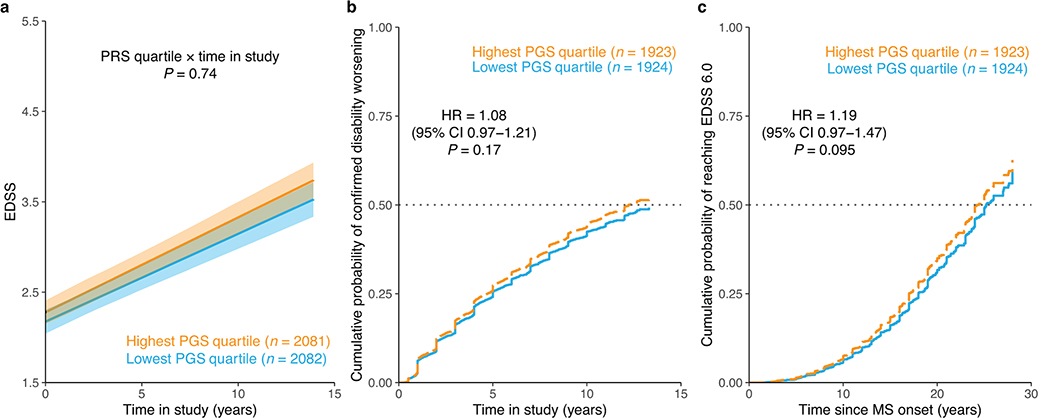
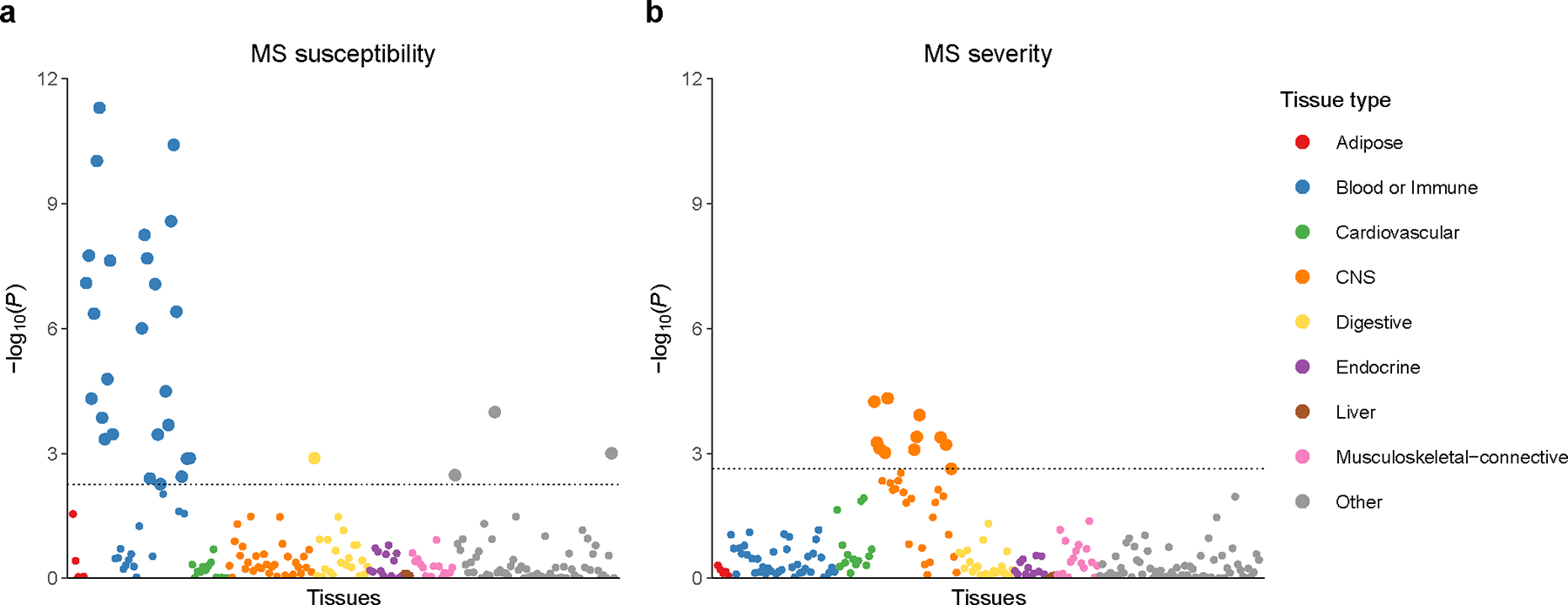
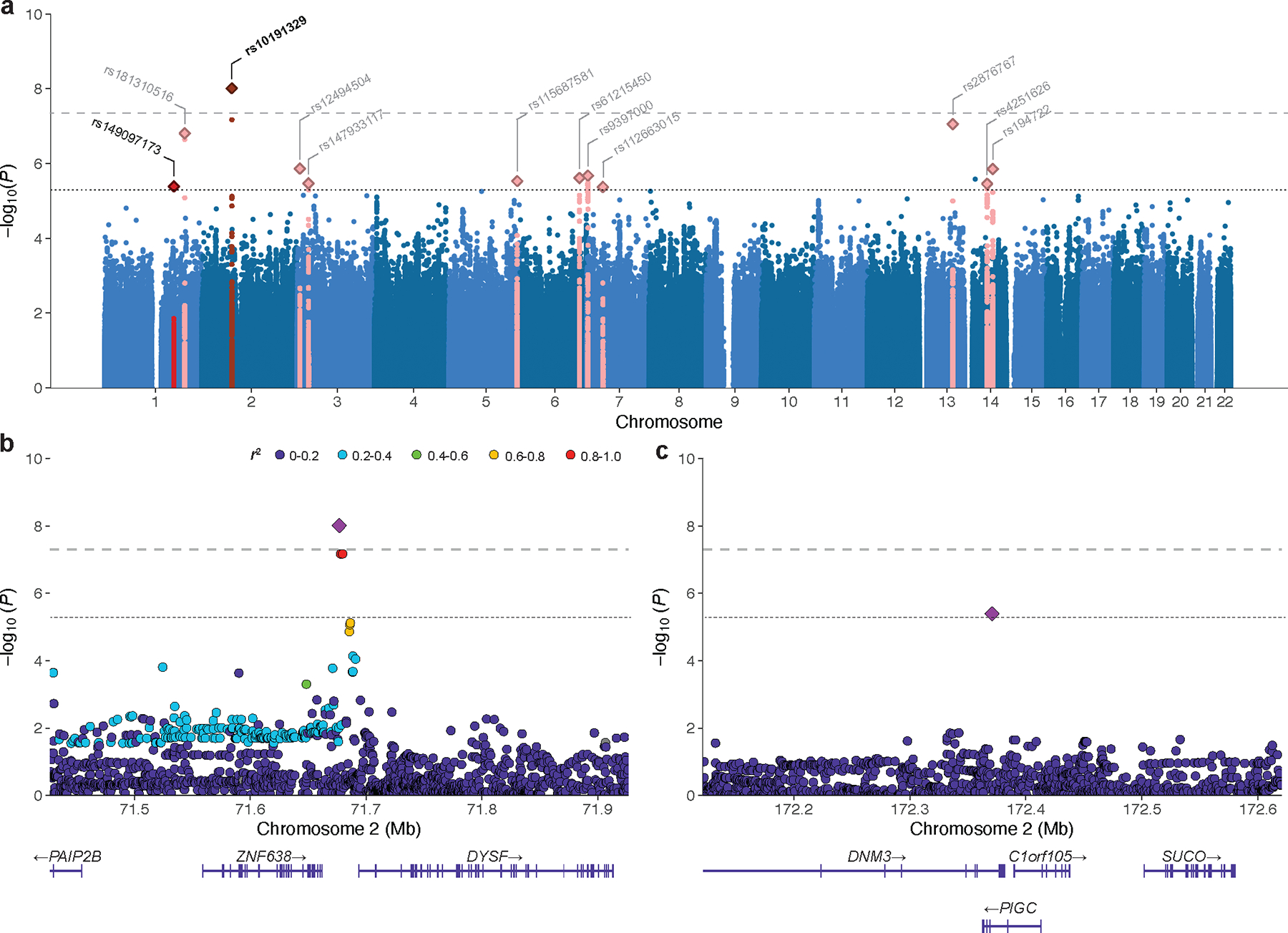
References
MAIN REFERENCES
-
- Thompson AJ, Baranzini SE, Geurts J, Hemmer B & Ciccarelli O Multiple sclerosis. Lancet 391, 1622–1636 (2018). - PubMed
METHODS REFERENCES
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
