

TURBOMOLE: Today and Tomorrow
- PMID: 37382508
- PMCID: PMC10601488
- DOI: 10.1021/acs.jctc.3c00347
TURBOMOLE: Today and Tomorrow
Abstract
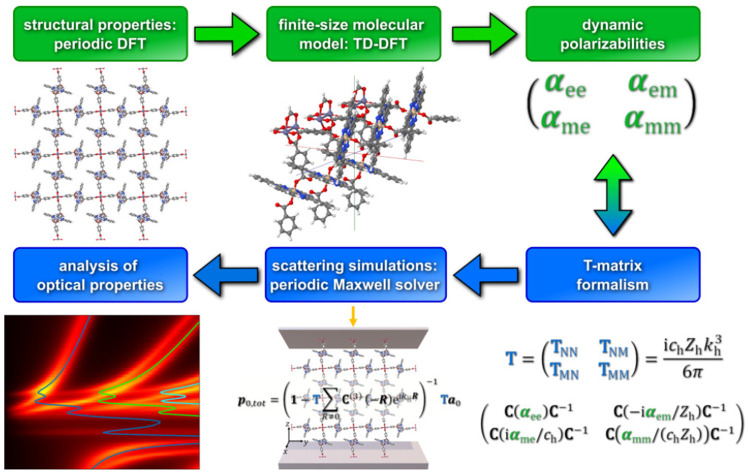
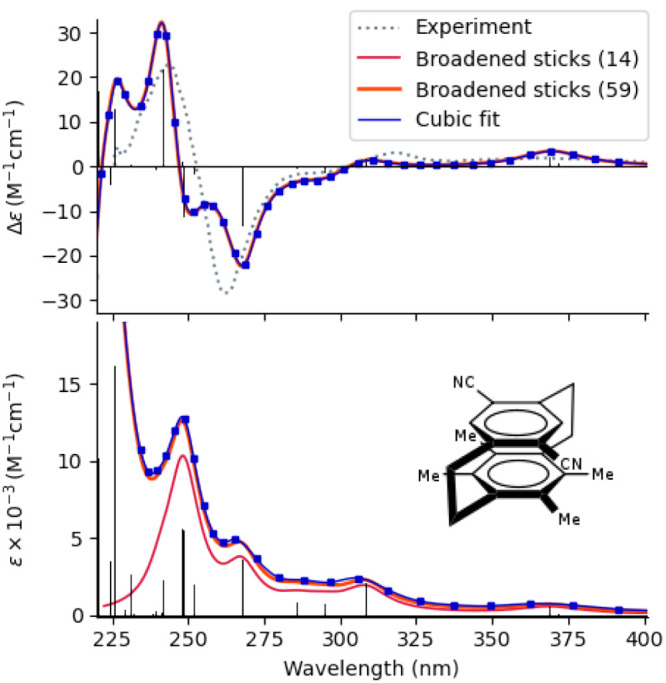
TURBOMOLE is a highly optimized software suite for large-scale quantum-chemical and materials science simulations of molecules, clusters, extended systems, and periodic solids. TURBOMOLE uses Gaussian basis sets and has been designed with robust and fast quantum-chemical applications in mind, ranging from homogeneous and heterogeneous catalysis to inorganic and organic chemistry and various types of spectroscopy, light-matter interactions, and biochemistry. This Perspective briefly surveys TURBOMOLE's functionality and highlights recent developments that have taken place between 2020 and 2023, comprising new electronic structure methods for molecules and solids, previously unavailable molecular properties, embedding, and molecular dynamics approaches. Select features under development are reviewed to illustrate the continuous growth of the program suite, including nuclear electronic orbital methods, Hartree-Fock-based adiabatic connection models, simplified time-dependent density functional theory, relativistic effects and magnetic properties, and multiscale modeling of optical properties.
Conflict of interest statement
The authors declare the following competing financial interest(s): Principal Investigator Filipp Furche has an equity interest in TURBOMOLE GmbH. The terms of this arrangement have been reviewed and approved by the University of California, Irvine, in accordance with its conflict of interest policies. Christof Hattig and David P. Tew have an equity interest in TURBOMOLE GmbH. Marek Sierka and Florian Weigend have an equity interest in TURBOMOLE GmbH and serve as its chief executive officers.
Figures
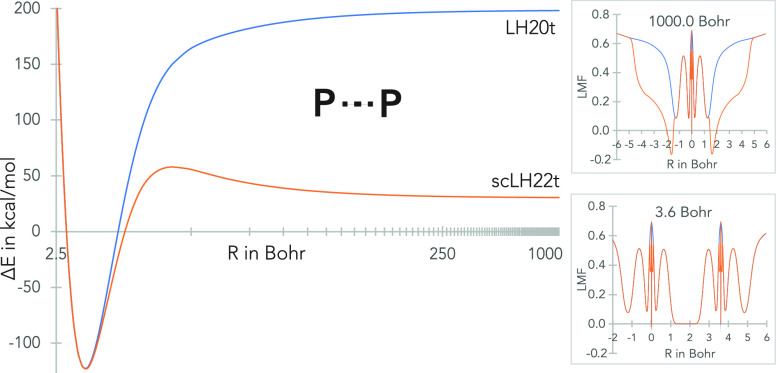
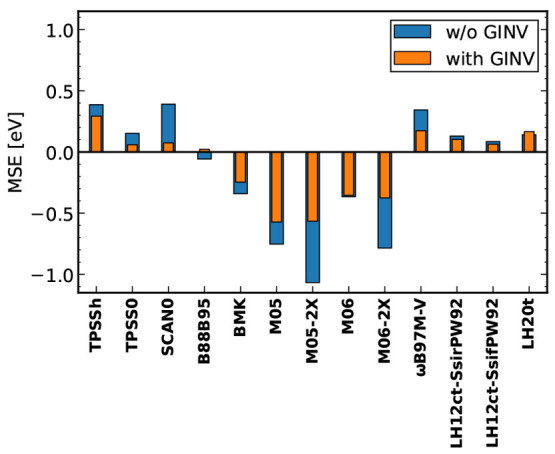
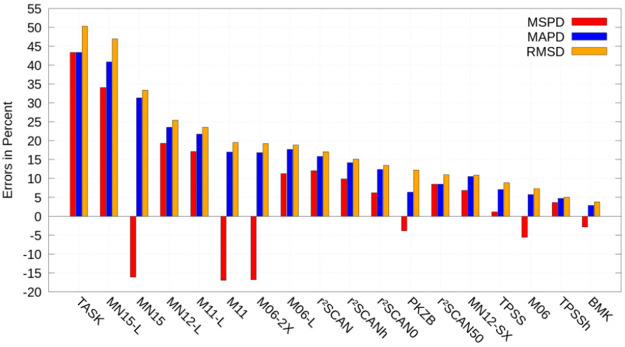
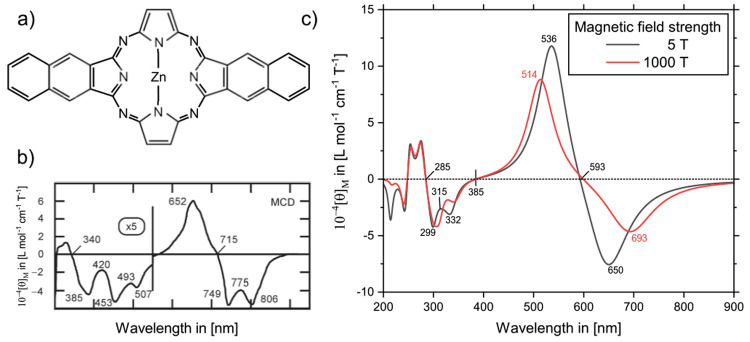
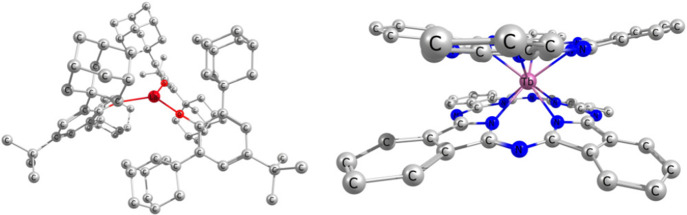
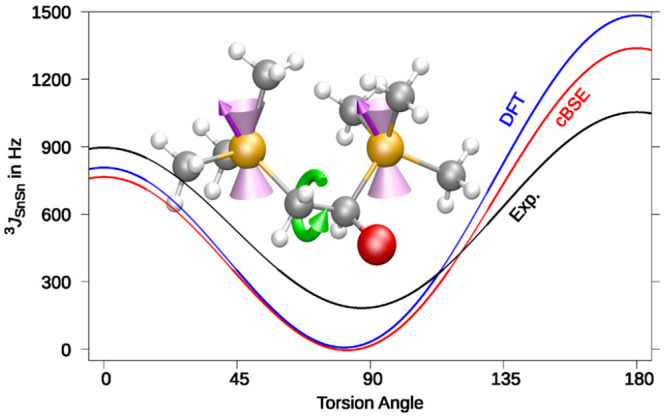
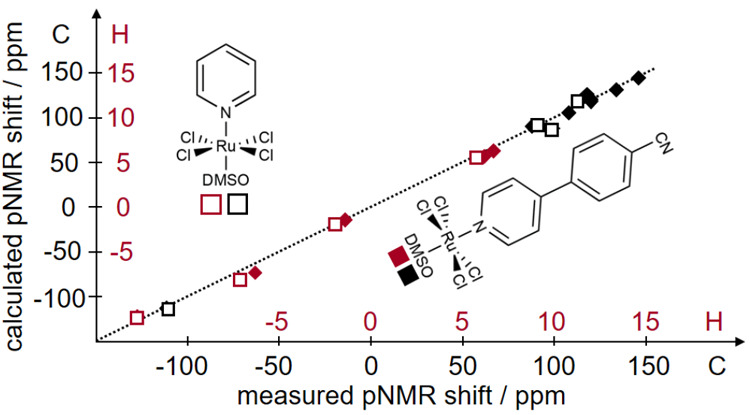
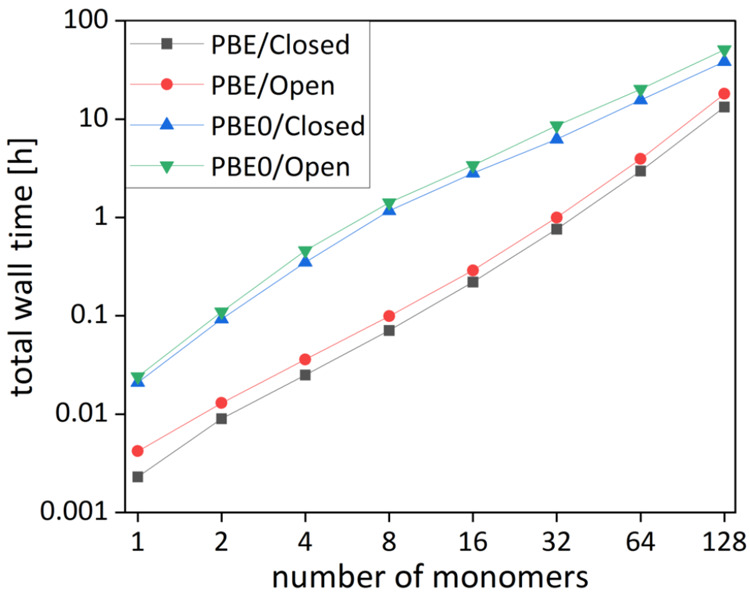

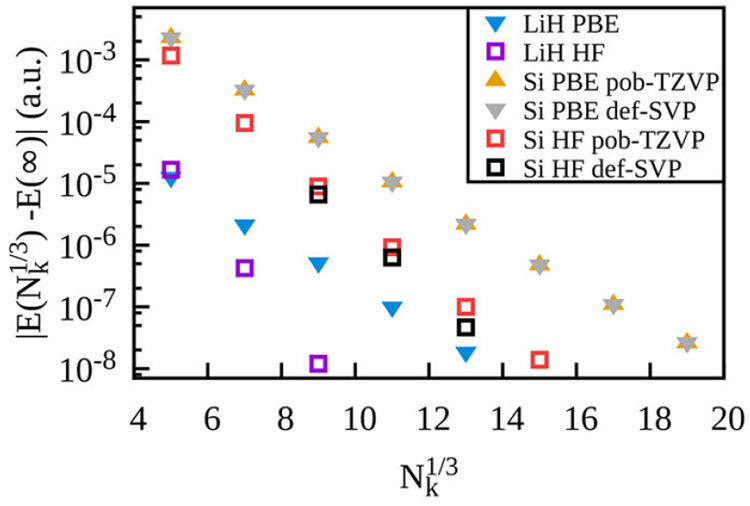
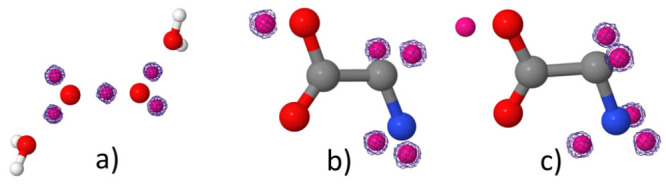
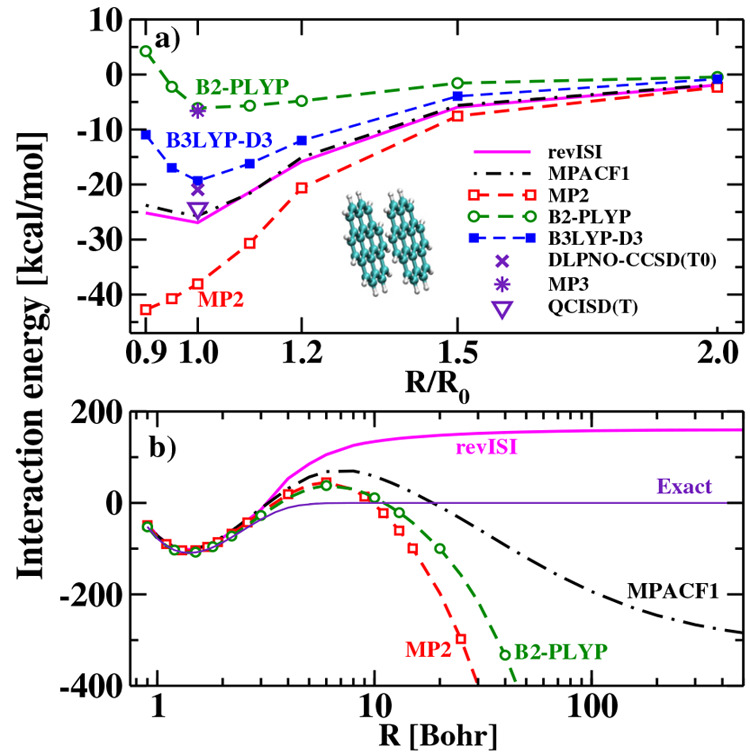
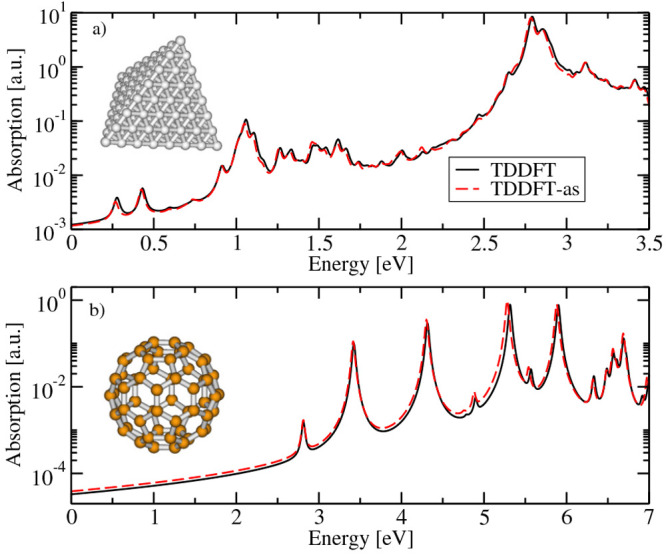
References
-
- Ahlrichs R.; Bär M.; Häser M.; Horn H.; Kölmel C. Electronic structure calculations on workstation computers: The program system TURBOMOLE. Chem. Phys. Lett. 1989, 162, 165–169. 10.1016/0009-2614(89)85118-8. - DOI
-
- Häser M.; Ahlrichs R. Improvements on the direct SCF method. J. Comput. Chem. 1989, 10, 104–111. 10.1002/jcc.540100111. - DOI
-
- Häser M.; Ahlrichs R.; Baron H. P.; Weis P.; Horn H. Direct computation of second-order SCF properties of large molecules on workstation computers with an application to large carbon clusters. Theor. Chim. Acta 1992, 83, 455–470. 10.1007/BF01113068. - DOI
-
- Haase F.; Ahlrichs R. Semidirect MP2 gradient evaluation on workstation computers: The MPGRAD program. J. Comput. Chem. 1993, 14, 907–912. 10.1002/jcc.540140805. - DOI
-
- Brode S.; Horn H.; Ehrig M.; Moldrup D.; Rice J. E.; Ahlrichs R. Parallel Direct SCF and Gradient Program For Workstation Clusters. J. Comput. Chem. 1993, 14, 1142–1148. 10.1002/jcc.540141004. - DOI
