

Single-cell multi-omics of mitochondrial DNA disorders reveals dynamics of purifying selection across human immune cells
- PMID: 37386249
- PMCID: PMC10548551
- DOI: 10.1038/s41588-023-01433-8
Single-cell multi-omics of mitochondrial DNA disorders reveals dynamics of purifying selection across human immune cells
Abstract
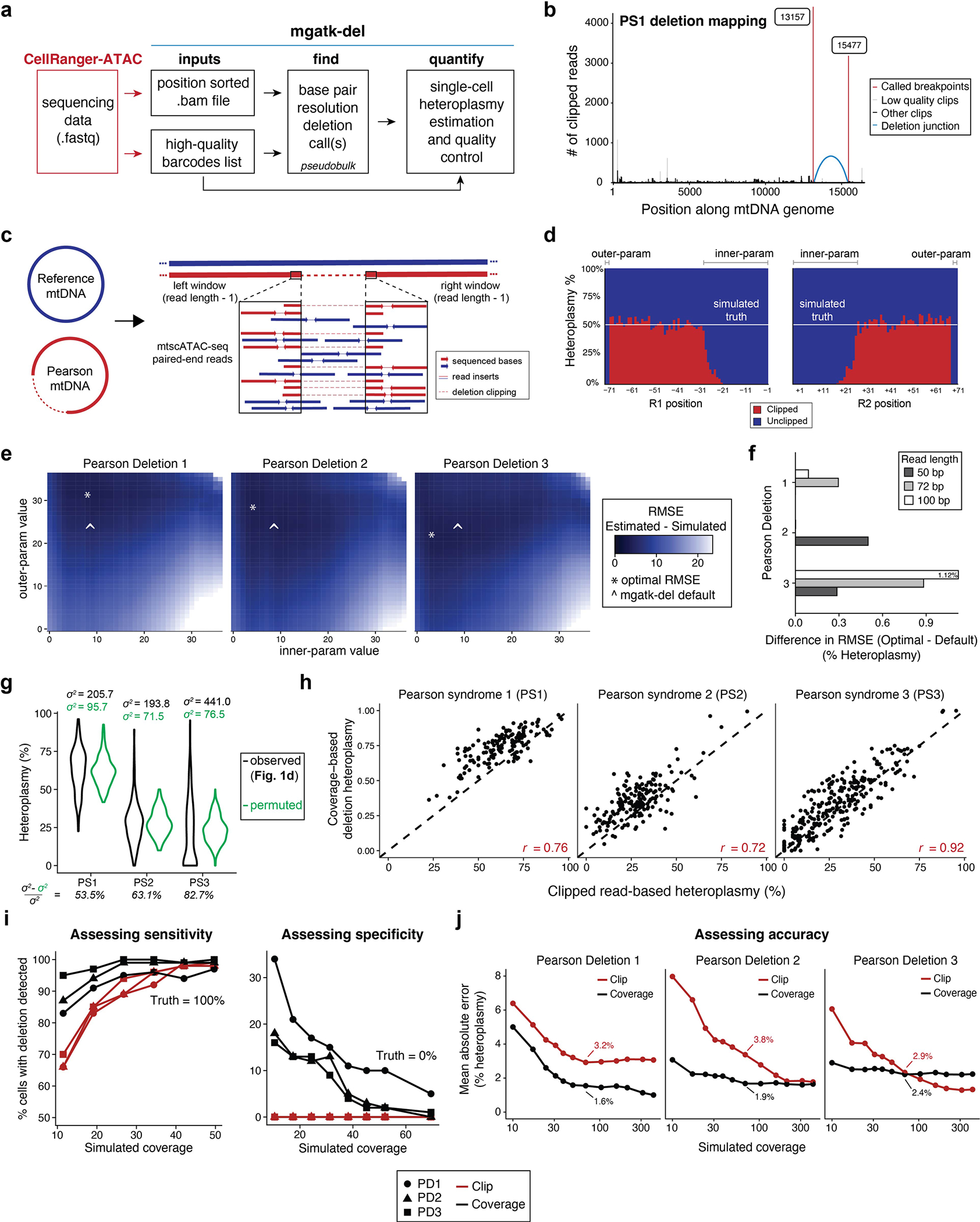
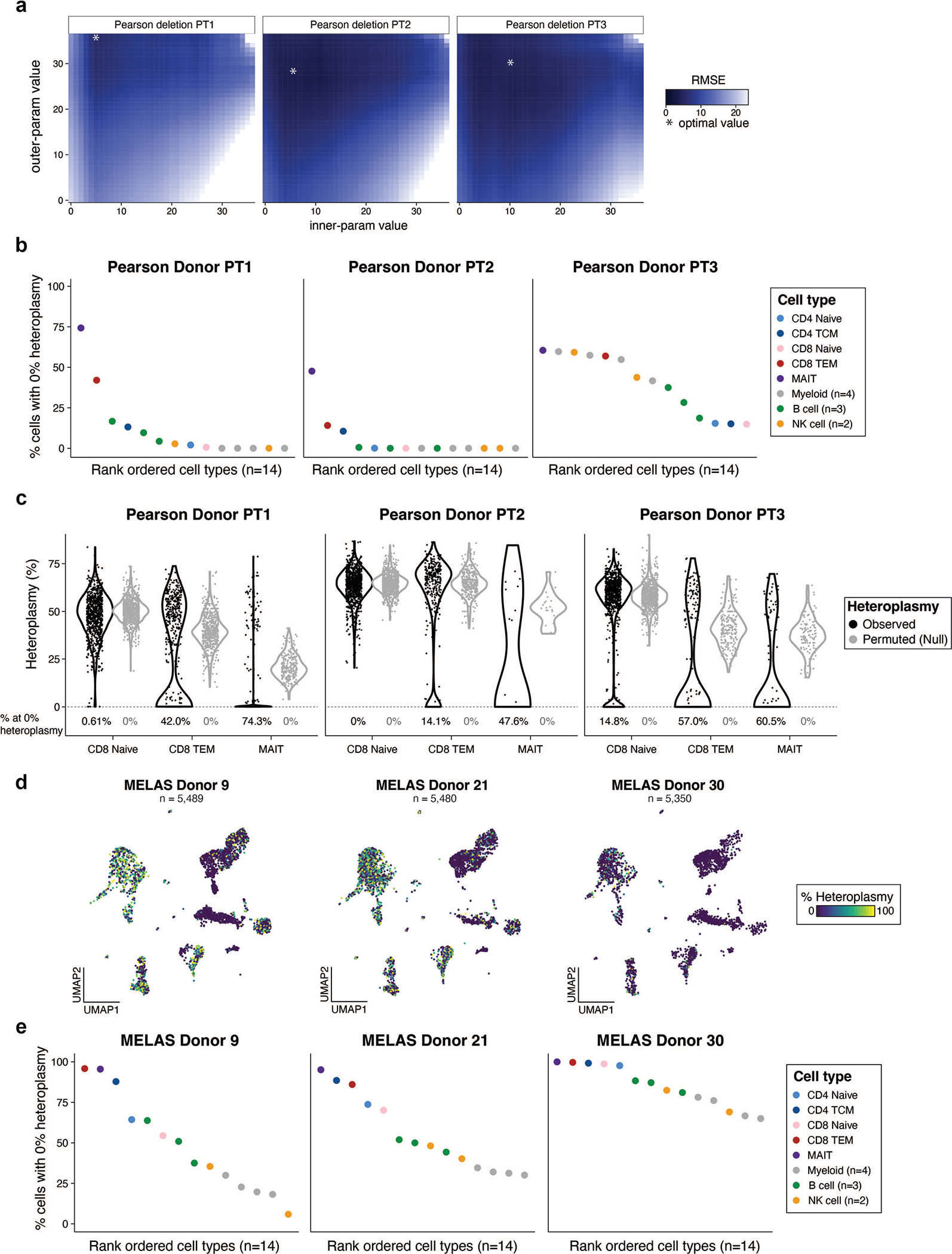
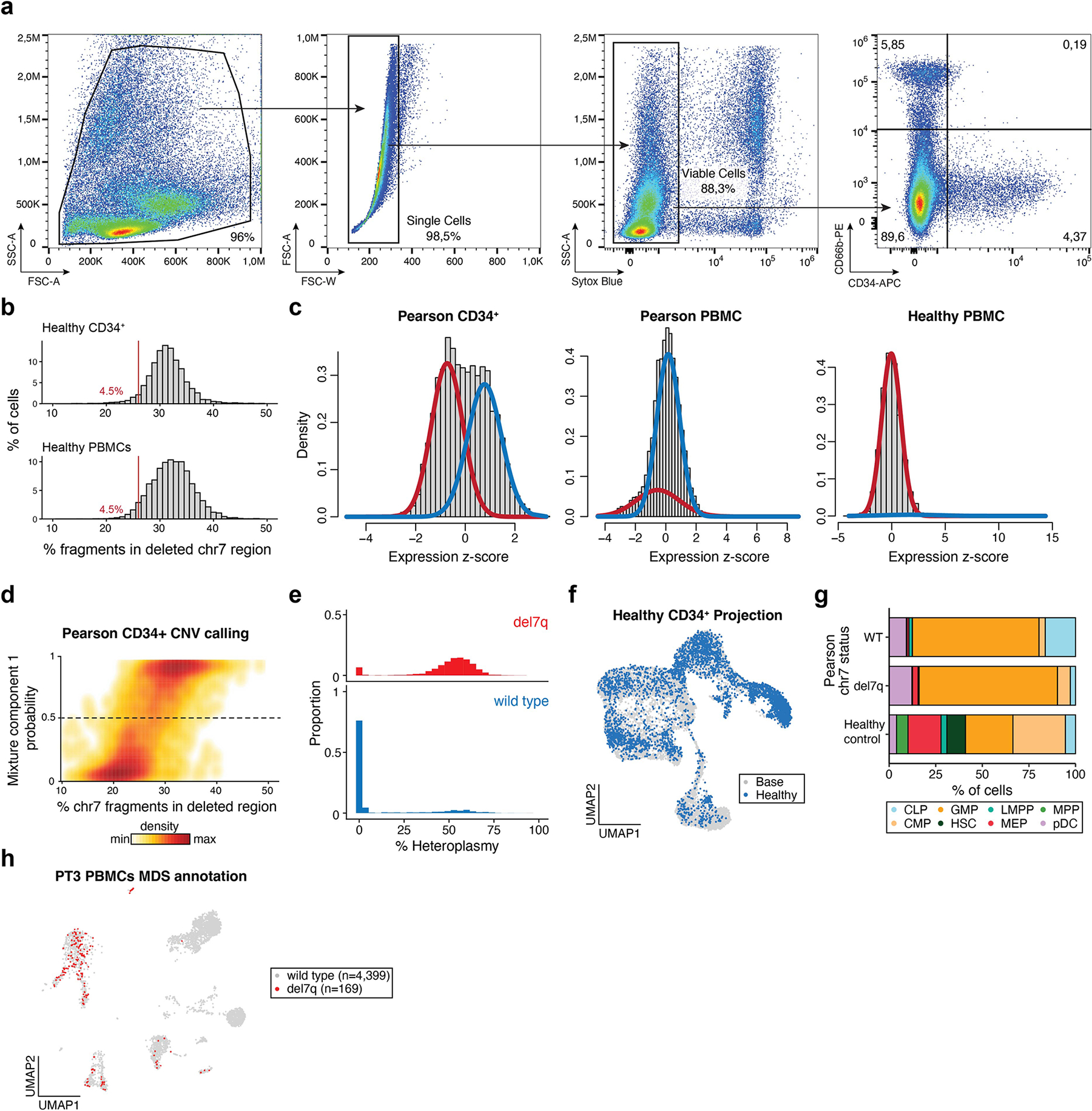
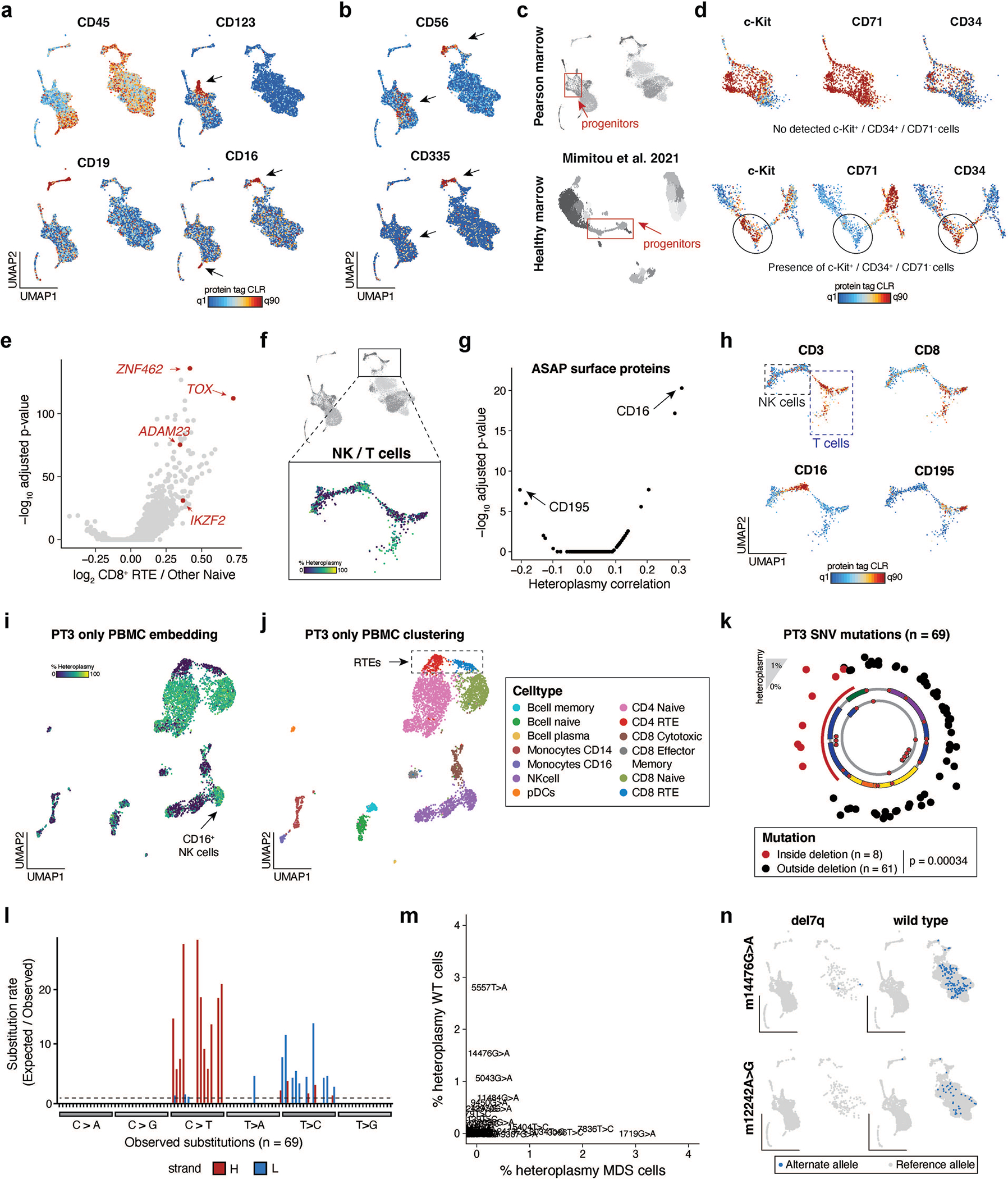
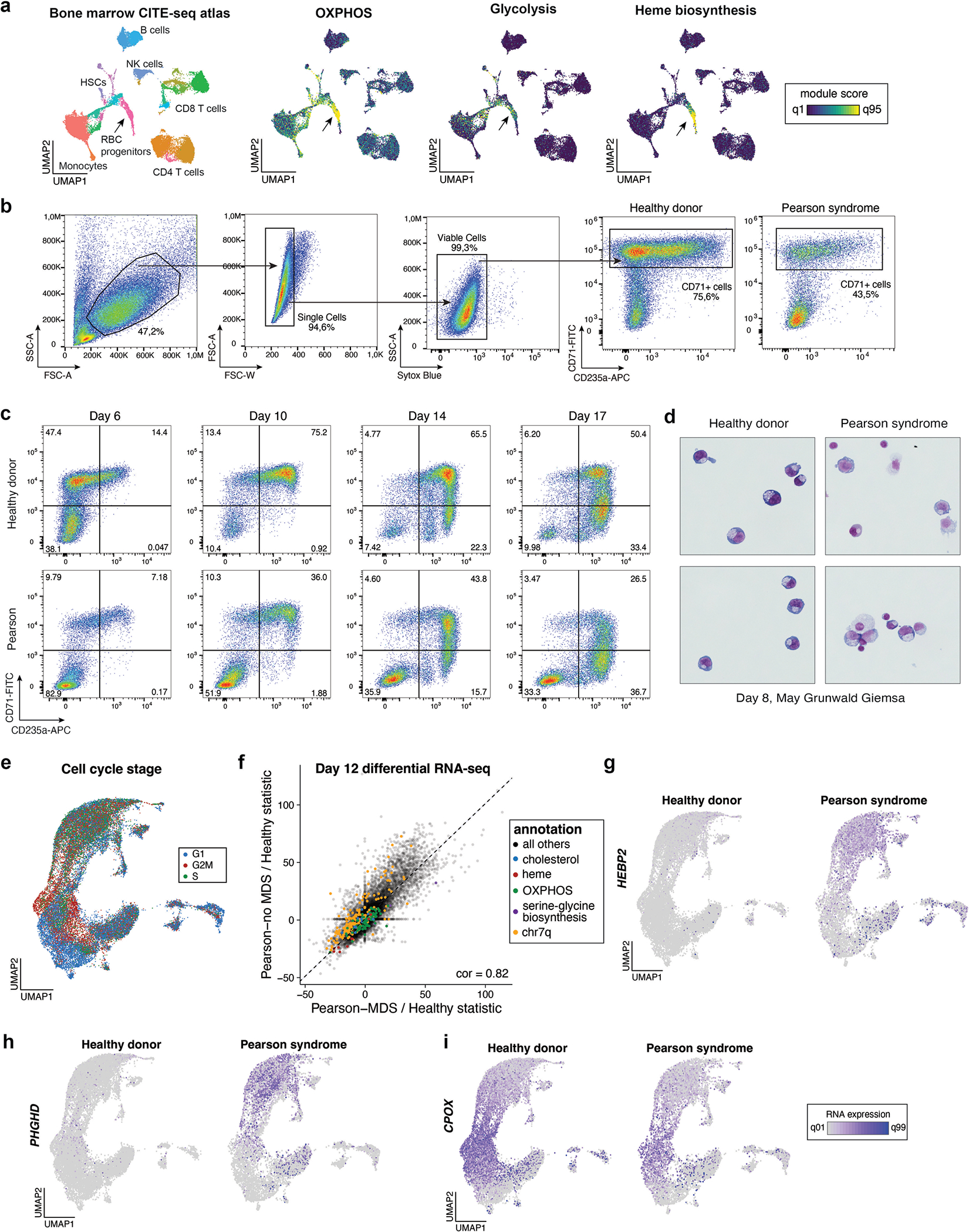
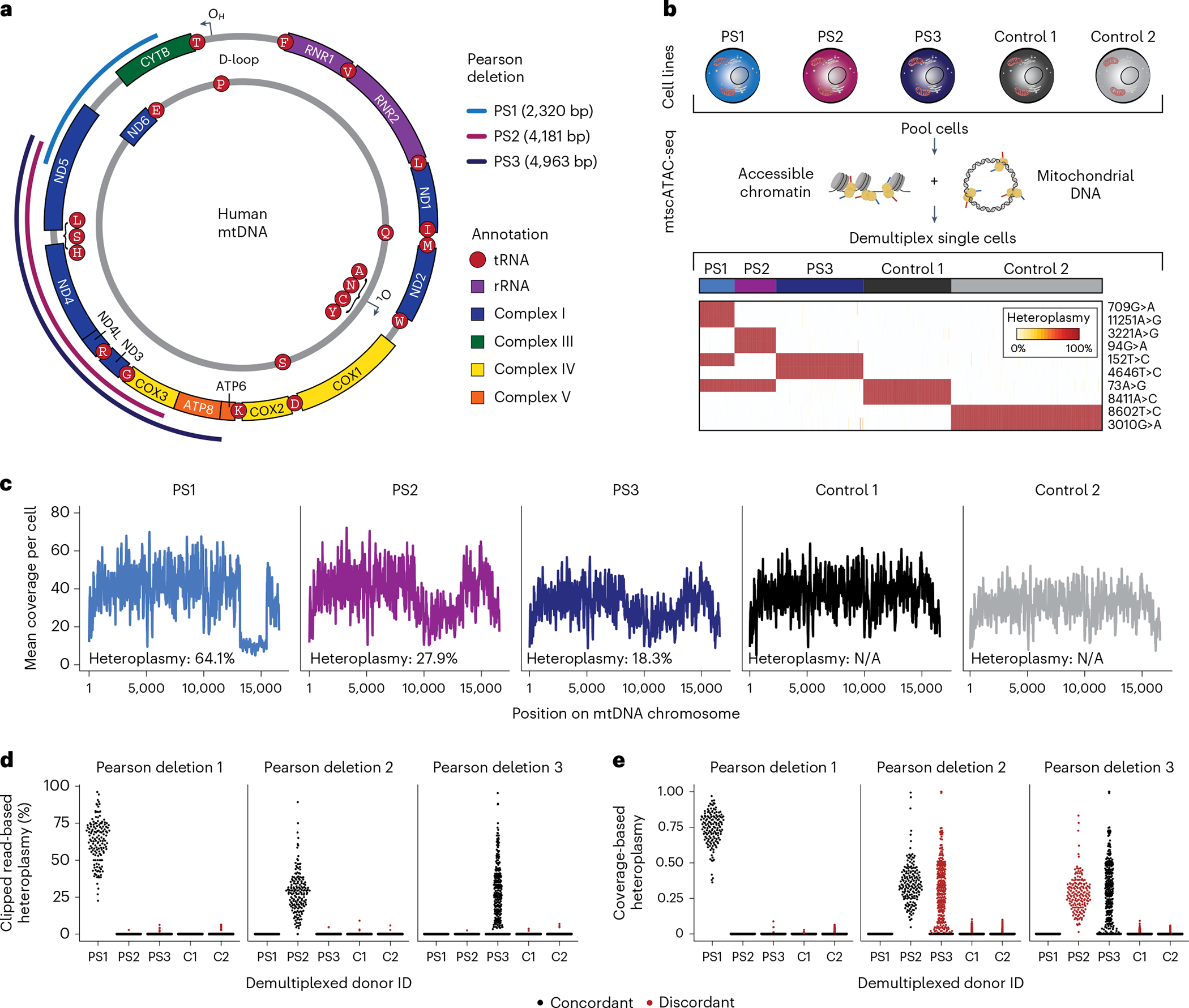
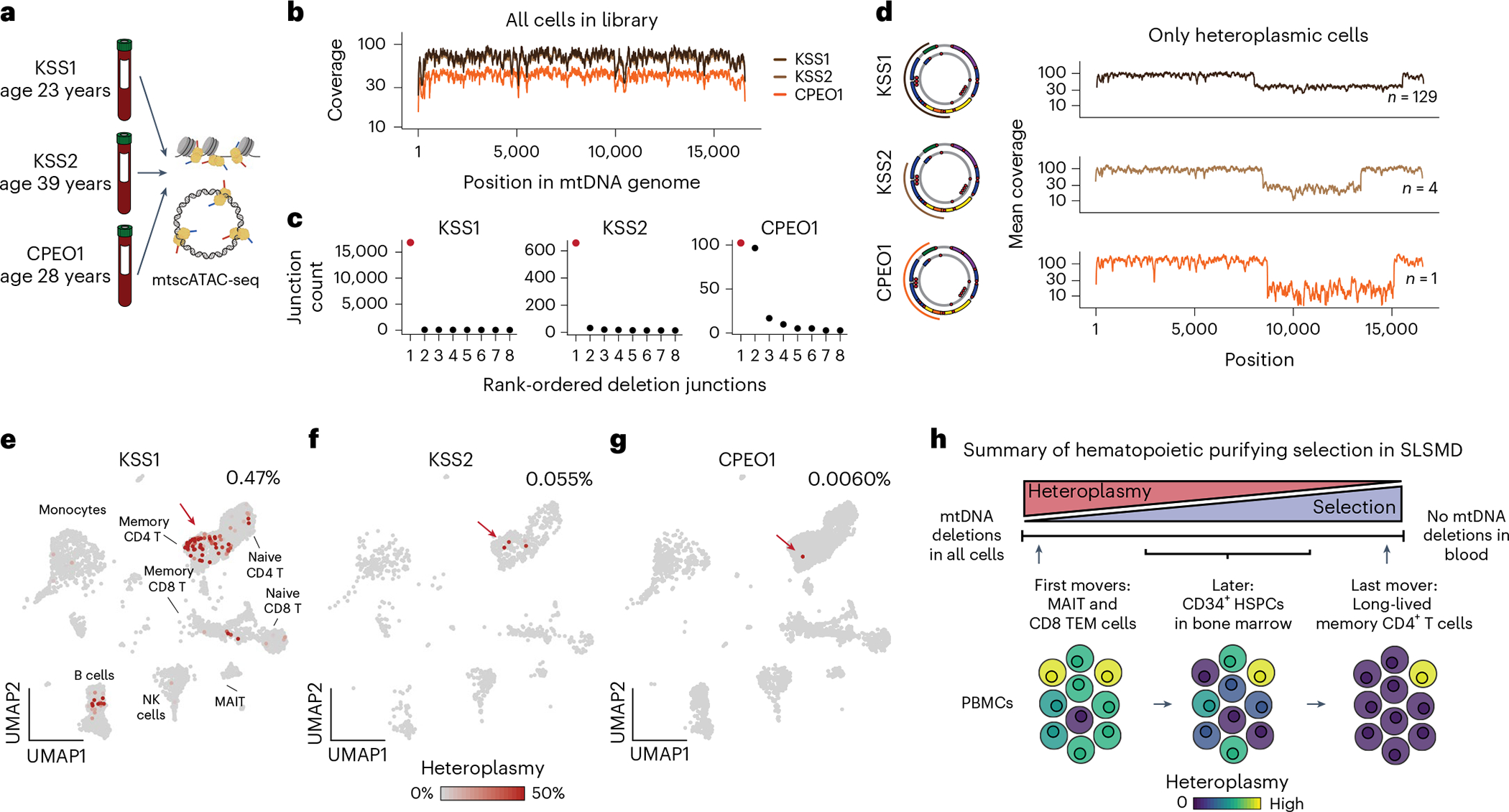
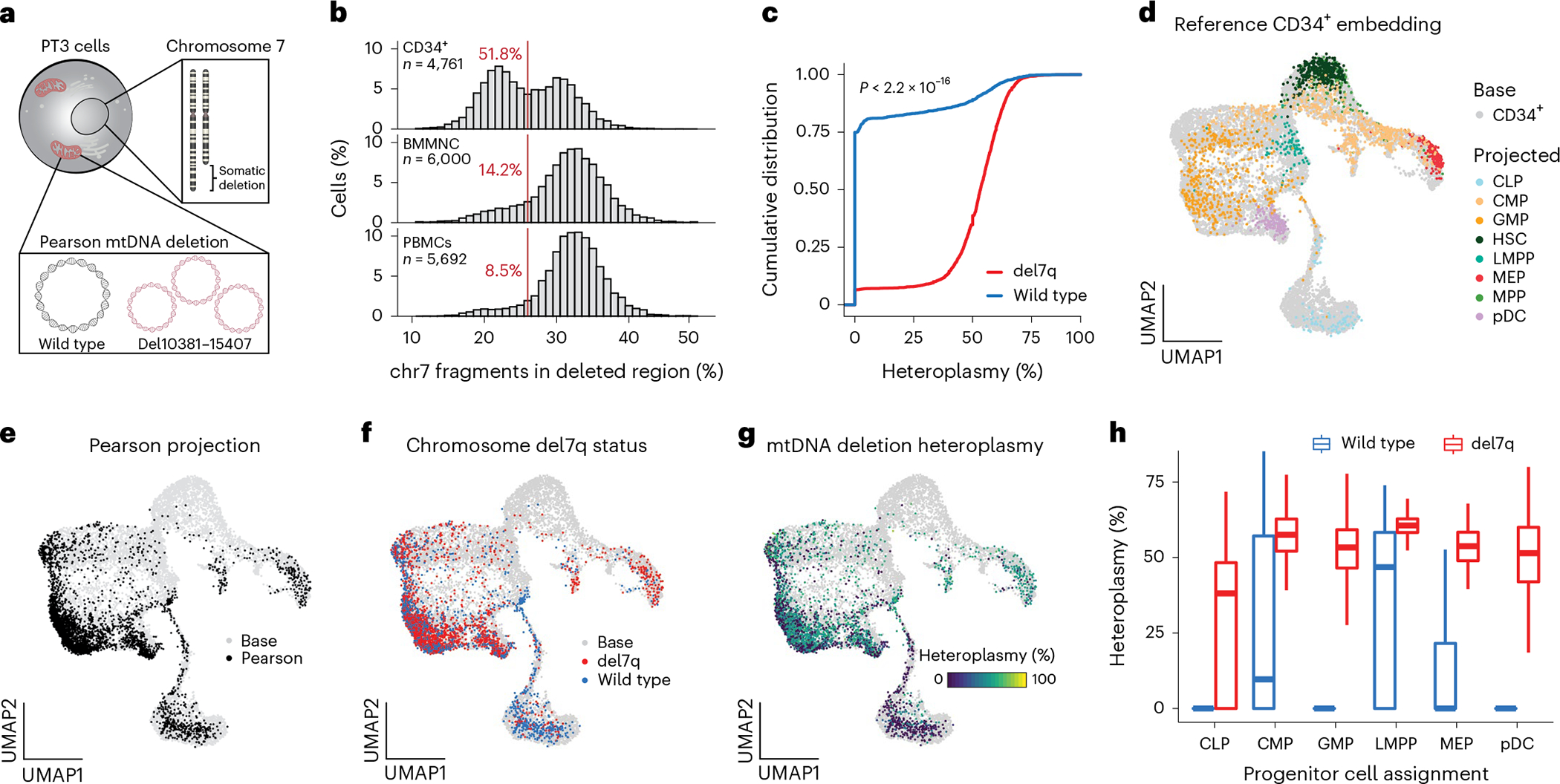
Pathogenic mutations in mitochondrial DNA (mtDNA) compromise cellular metabolism, contributing to cellular heterogeneity and disease. Diverse mutations are associated with diverse clinical phenotypes, suggesting distinct organ- and cell-type-specific metabolic vulnerabilities. Here we establish a multi-omics approach to quantify deletions in mtDNA alongside cell state features in single cells derived from six patients across the phenotypic spectrum of single large-scale mtDNA deletions (SLSMDs). By profiling 206,663 cells, we reveal the dynamics of pathogenic mtDNA deletion heteroplasmy consistent with purifying selection and distinct metabolic vulnerabilities across T-cell states in vivo and validate these observations in vitro. By extending analyses to hematopoietic and erythroid progenitors, we reveal mtDNA dynamics and cell-type-specific gene regulatory adaptations, demonstrating the context-dependence of perturbing mitochondrial genomic integrity. Collectively, we report pathogenic mtDNA heteroplasmy dynamics of individual blood and immune cells across lineages, demonstrating the power of single-cell multi-omics for revealing fundamental properties of mitochondrial genetics.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
The Broad Institute has filed for a patent relating to the use of the technology described in this paper where C.A.L., L.S.L., C.M., A.R. and V.G.S. are named inventors (US provisional patent application 62/683,502). C.A.L. and L.S.L. are consultants to Cartography Biosciences. ATS is a founder of Immunai and Cartography Biosciences and receives research funding from Allogene Therapeutics and Merck Research Laboratories. A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics, and until August 31, 2020, was an SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and ThermoFisher Scientific. Since August 1, 2020, A.R. has been an employee of Genentech. V.G.S. serves as an advisor to and/or has equity in Branch Biosciences, Novartis, Forma, Cellarity and Ensoma. S.A.V. is an advisor to Immunai and has provided consulting services to Koch Disruptive Technologies and ADC Therapeutics. The remaining authors declare no competing interests.
Figures




Comment in
-
Single-cell multiomic analyses sheds light on mitochondrial mutational selection.Nat Genet. 2023 Jul;55(7):1083-1085. doi: 10.1038/s41588-023-01436-5. Nat Genet. 2023. PMID: 37386252 No abstract available.
References
-
- Stewart JB & Chinnery PF Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat. Rev. Genet. 22, 106–118 (2021). - PubMed
-
- Stewart JB & Chinnery PF The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- UM1 HG012076/HG/NHGRI NIH HHS/United States
- R33 HL154133/HL/NHLBI NIH HHS/United States
- T32 HL007574/HL/NHLBI NIH HHS/United States
- R00 HG012579/HG/NHGRI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- U54 NS078059/NS/NINDS NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R33 HL120791/HL/NHLBI NIH HHS/United States
- K99 HG012579/HG/NHGRI NIH HHS/United States
- RC2 DK122533/DK/NIDDK NIH HHS/United States
- R01 DK103794/DK/NIDDK NIH HHS/United States
- R01 DK107716/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
