

Resolving therapy resistance mechanisms in multiple myeloma by multiomics subclone analysis
- PMID: 37390336
- PMCID: PMC10733835
- DOI: 10.1182/blood.2023019758
Resolving therapy resistance mechanisms in multiple myeloma by multiomics subclone analysis
Abstract
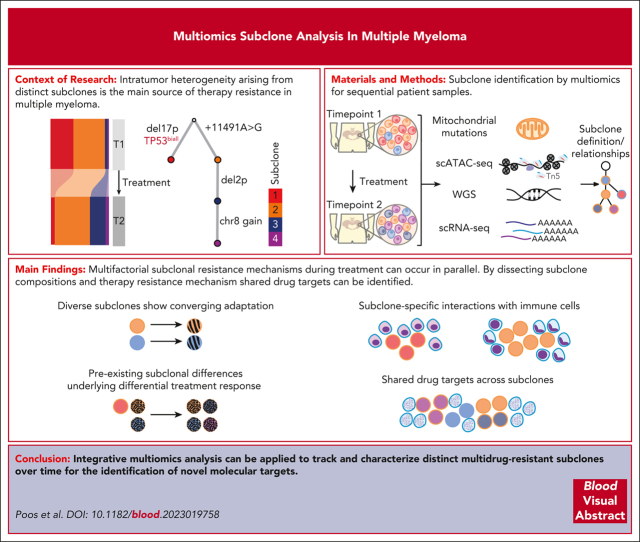
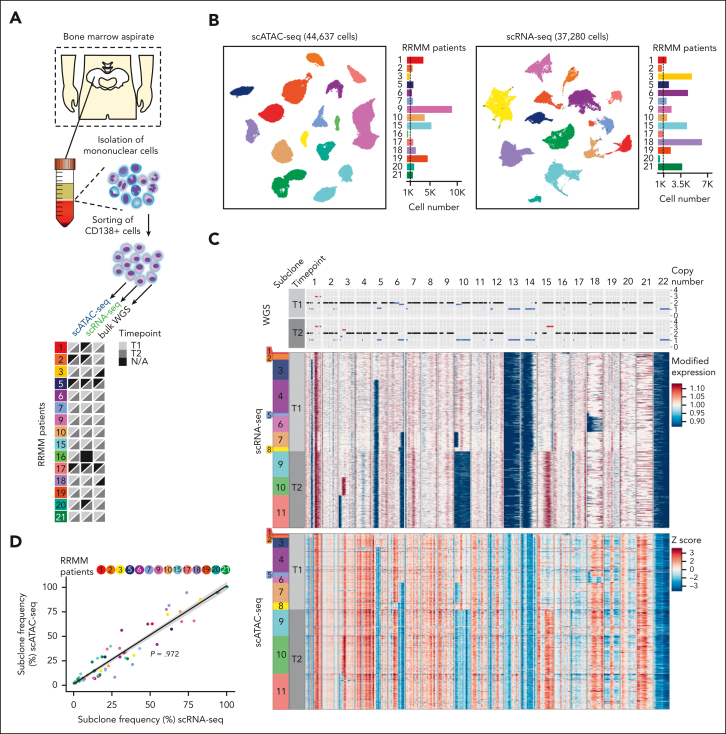
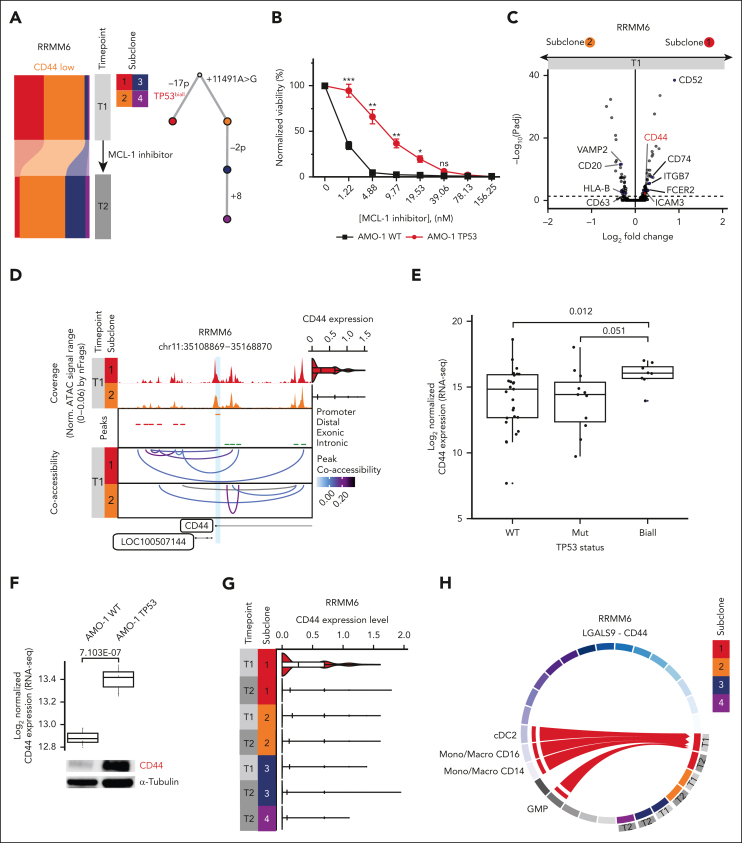
Intratumor heterogeneity as a clinical challenge becomes most evident after several treatment lines, when multidrug-resistant subclones accumulate. To address this challenge, the characterization of resistance mechanisms at the subclonal level is key to identify common vulnerabilities. In this study, we integrate whole-genome sequencing, single-cell (sc) transcriptomics (scRNA sequencing), and chromatin accessibility (scATAC sequencing) together with mitochondrial DNA mutations to define subclonal architecture and evolution for longitudinal samples from 15 patients with relapsed or refractory multiple myeloma. We assess transcriptomic and epigenomic changes to resolve the multifactorial nature of therapy resistance and relate it to the parallel occurrence of different mechanisms: (1) preexisting epigenetic profiles of subclones associated with survival advantages, (2) converging phenotypic adaptation of genetically distinct subclones, and (3) subclone-specific interactions of myeloma and bone marrow microenvironment cells. Our study showcases how an integrative multiomics analysis can be applied to track and characterize distinct multidrug-resistant subclones over time for the identification of molecular targets against them.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures




Comment in
-
Myeloma heterogeneity at cell resolution.Blood. 2023 Nov 9;142(19):1582-1583. doi: 10.1182/blood.2023021523. Blood. 2023. PMID: 37944180 No abstract available.
References
-
- Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335–348. - PubMed
-
- Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14(2):100–113. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
