

Fine-tuning GPCR-mediated neuromodulation by biasing signaling through different G protein subunits
- PMID: 37390816
- PMCID: PMC10527995
- DOI: 10.1016/j.molcel.2023.06.006
Fine-tuning GPCR-mediated neuromodulation by biasing signaling through different G protein subunits
Abstract
G-protein-coupled receptors (GPCRs) mediate neuromodulation through the activation of heterotrimeric G proteins (Gαβγ). Classical models depict that G protein activation leads to a one-to-one formation of Gα-GTP and Gβγ species. Each of these species propagates signaling by independently acting on effectors, but the mechanisms by which response fidelity is ensured by coordinating Gα and Gβγ responses remain unknown. Here, we reveal a paradigm of G protein regulation whereby the neuronal protein GINIP (Gα inhibitory interacting protein) biases inhibitory GPCR responses to favor Gβγ over Gα signaling. Tight binding of GINIP to Gαi-GTP precludes its association with effectors (adenylyl cyclase) and, simultaneously, with regulator-of-G-protein-signaling (RGS) proteins that accelerate deactivation. As a consequence, Gαi-GTP signaling is dampened, whereas Gβγ signaling is enhanced. We show that this mechanism is essential to prevent the imbalances of neurotransmission that underlie increased seizure susceptibility in mice. Our findings reveal an additional layer of regulation within a quintessential mechanism of signal transduction that sets the tone of neurotransmission.
Keywords: G protein; GPCR; GTPase; KIAA1045; PHF24; neuron; neurotransmitter; seizures.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
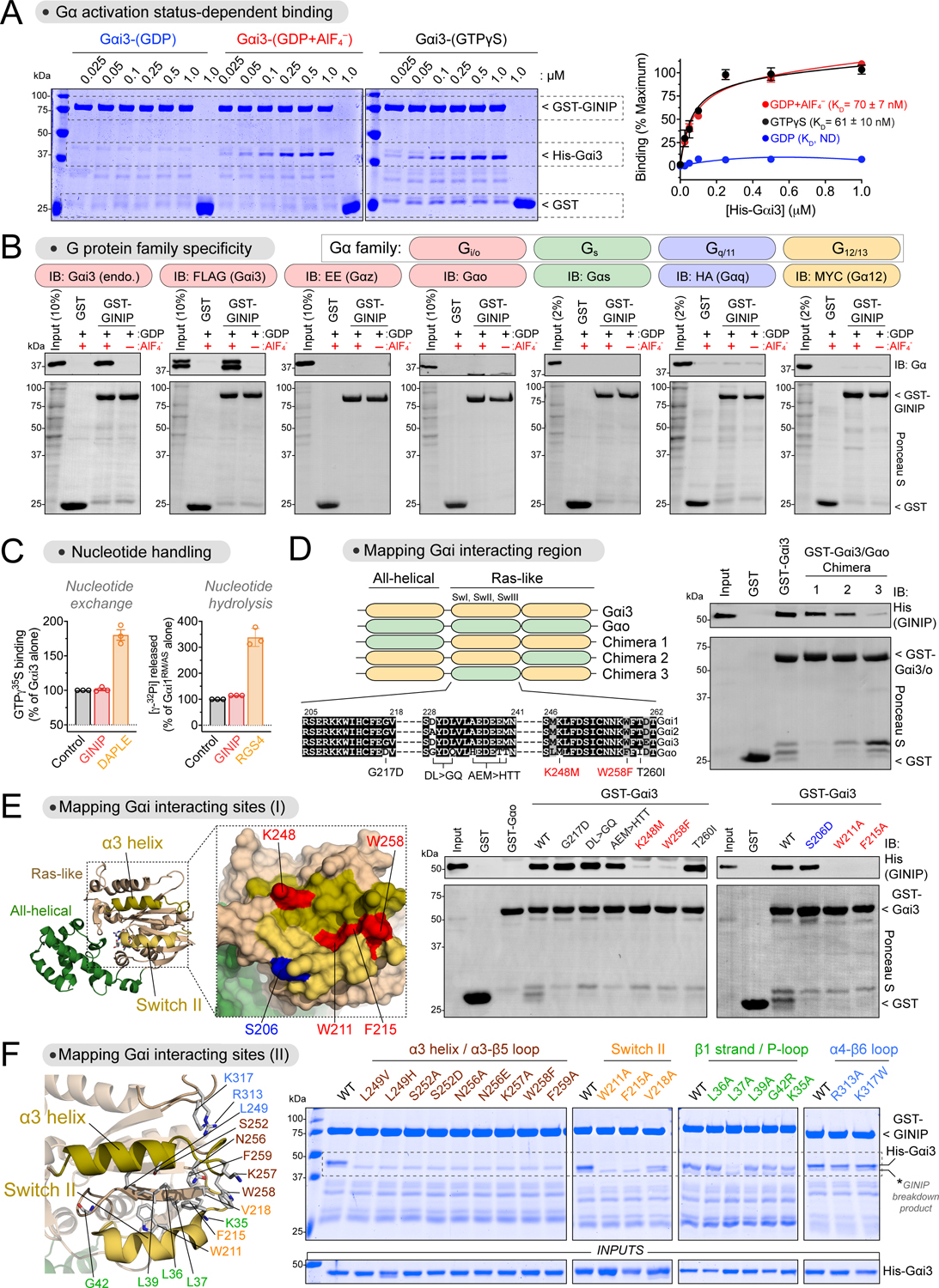
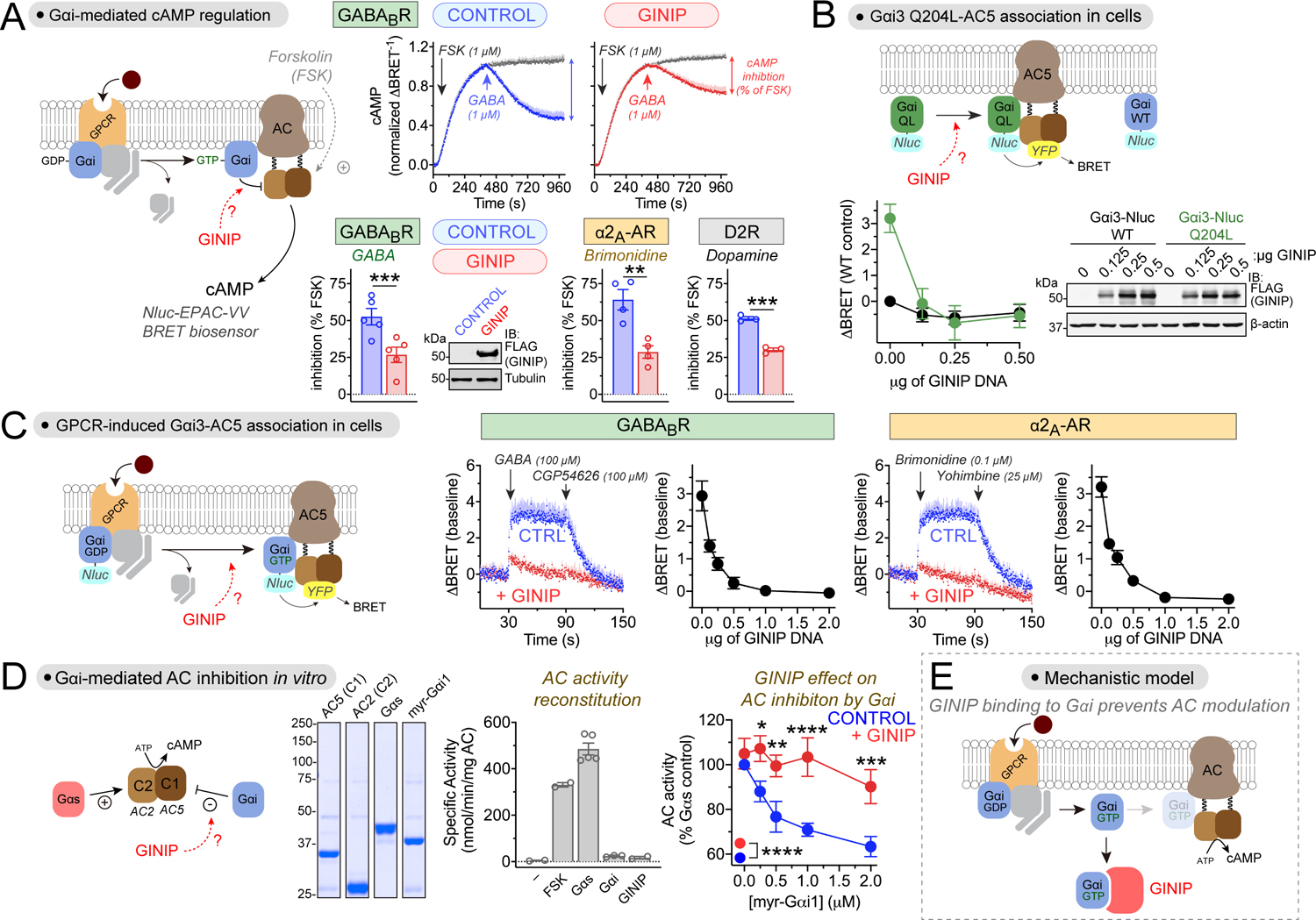
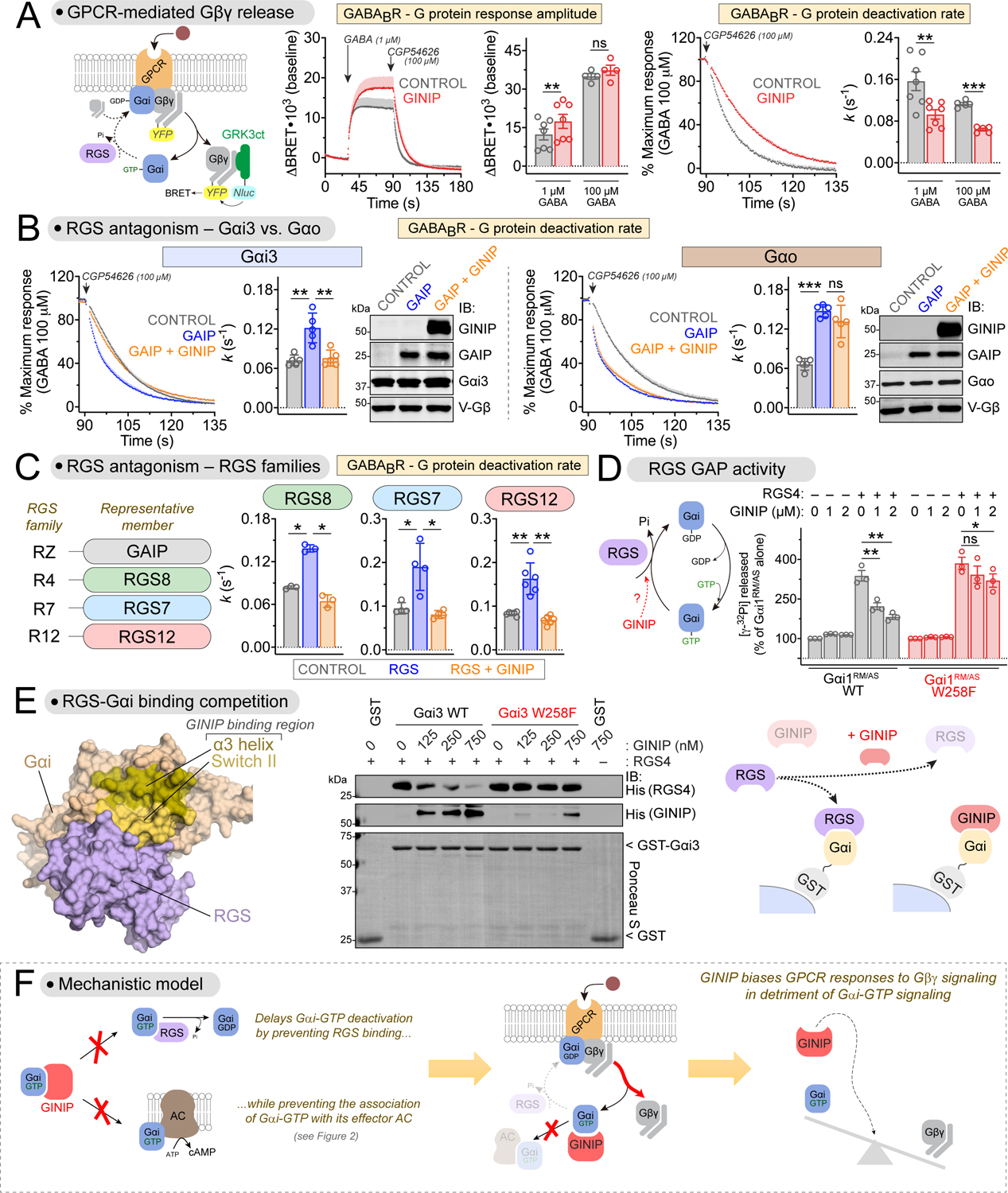
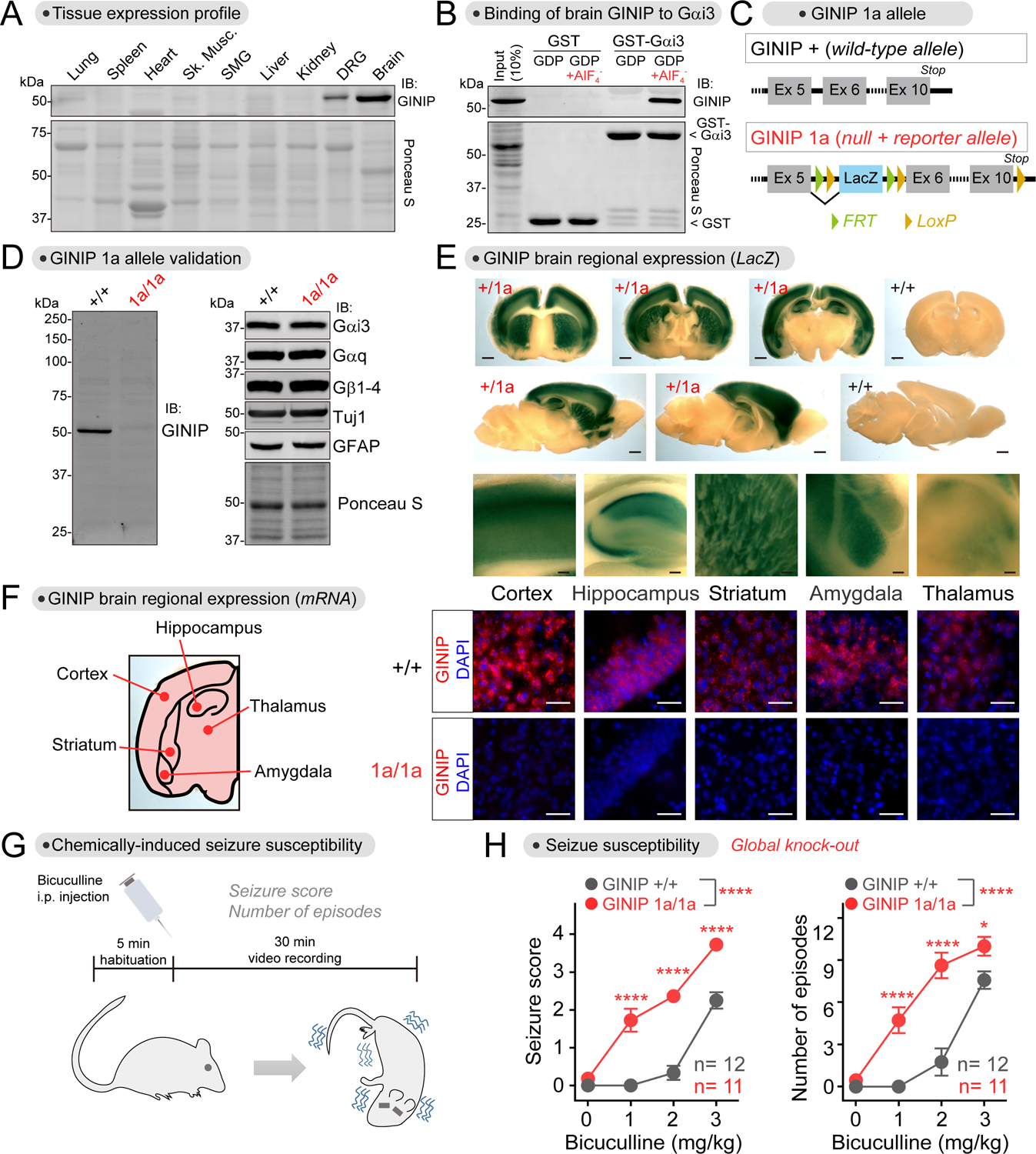
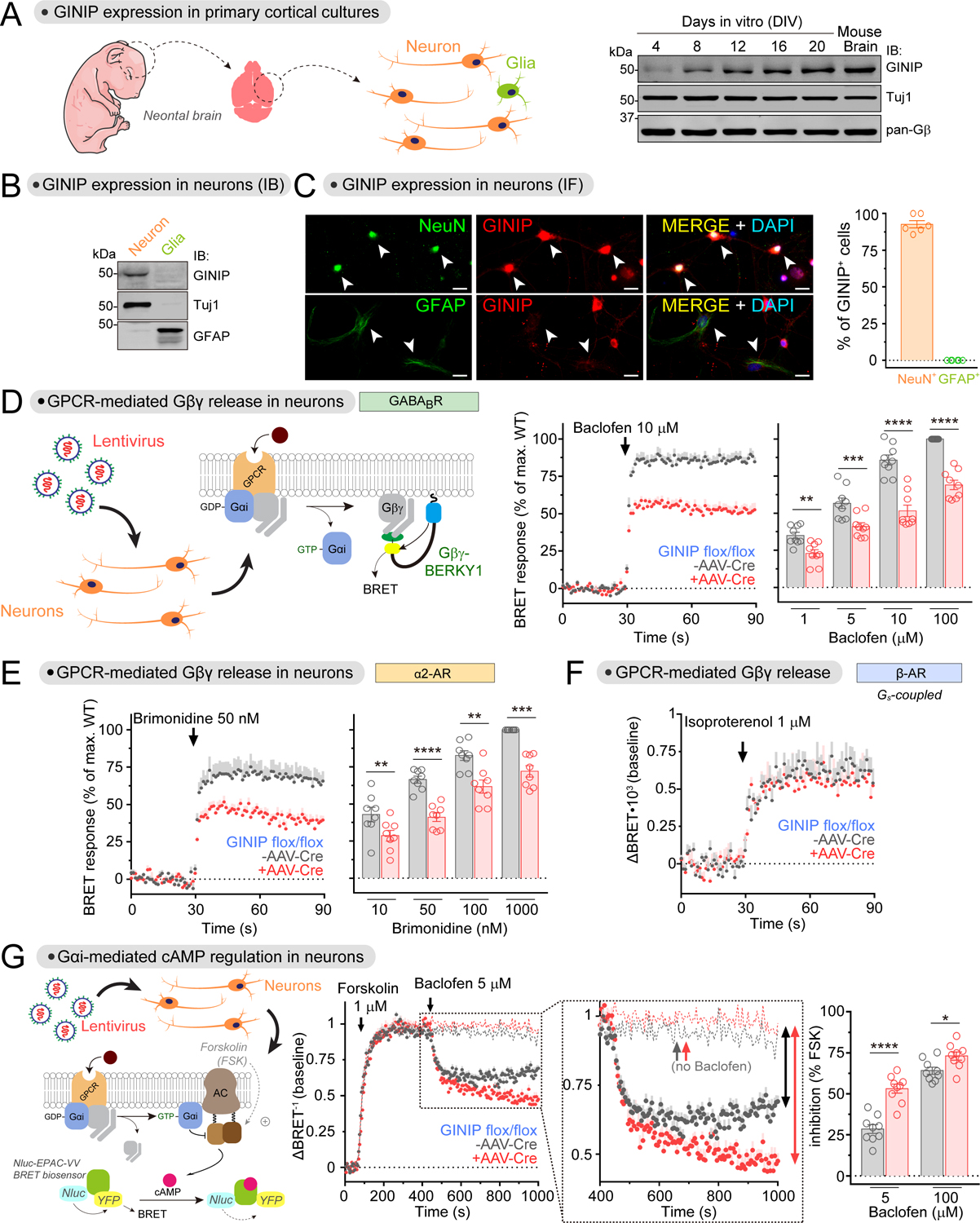
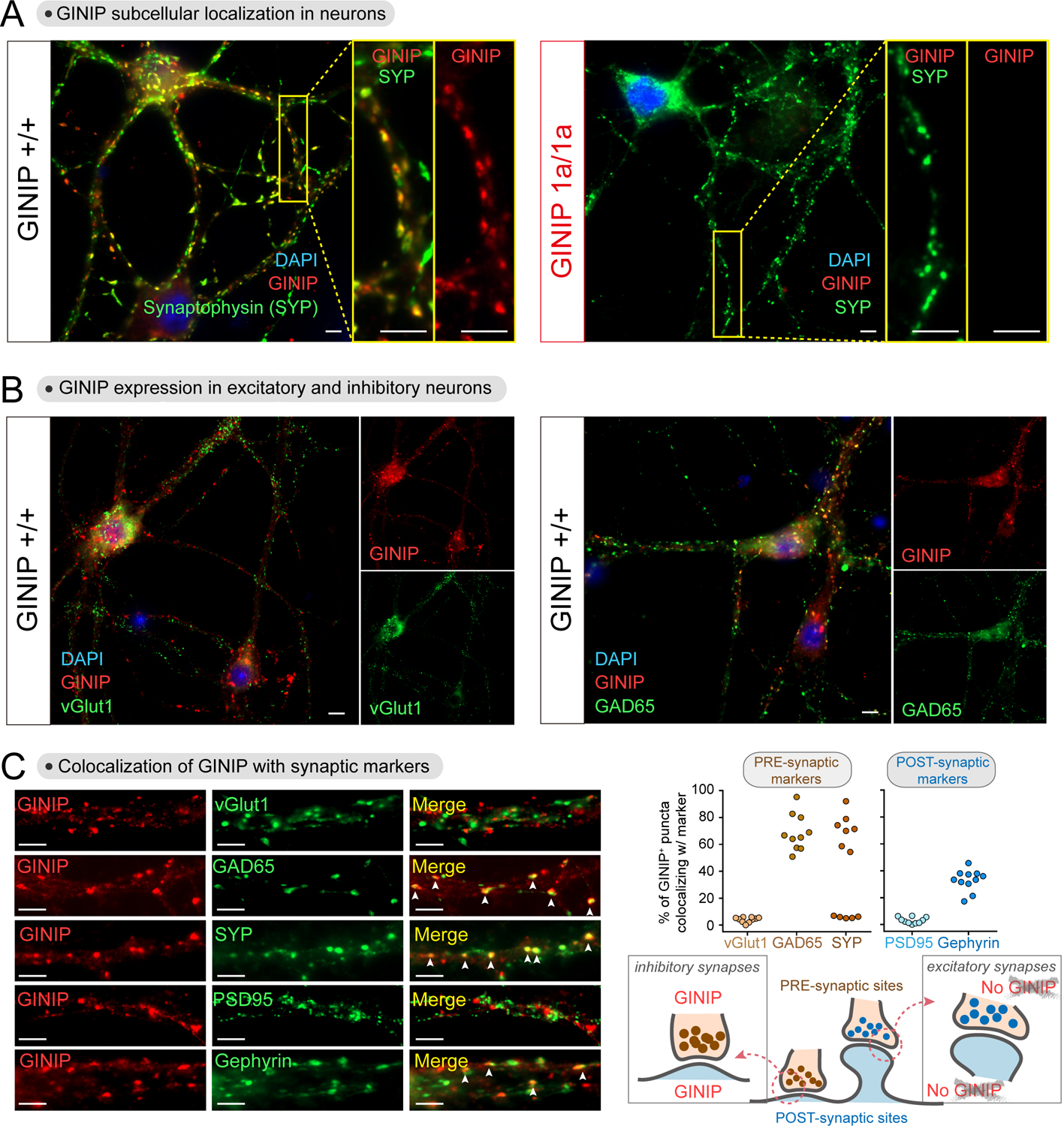
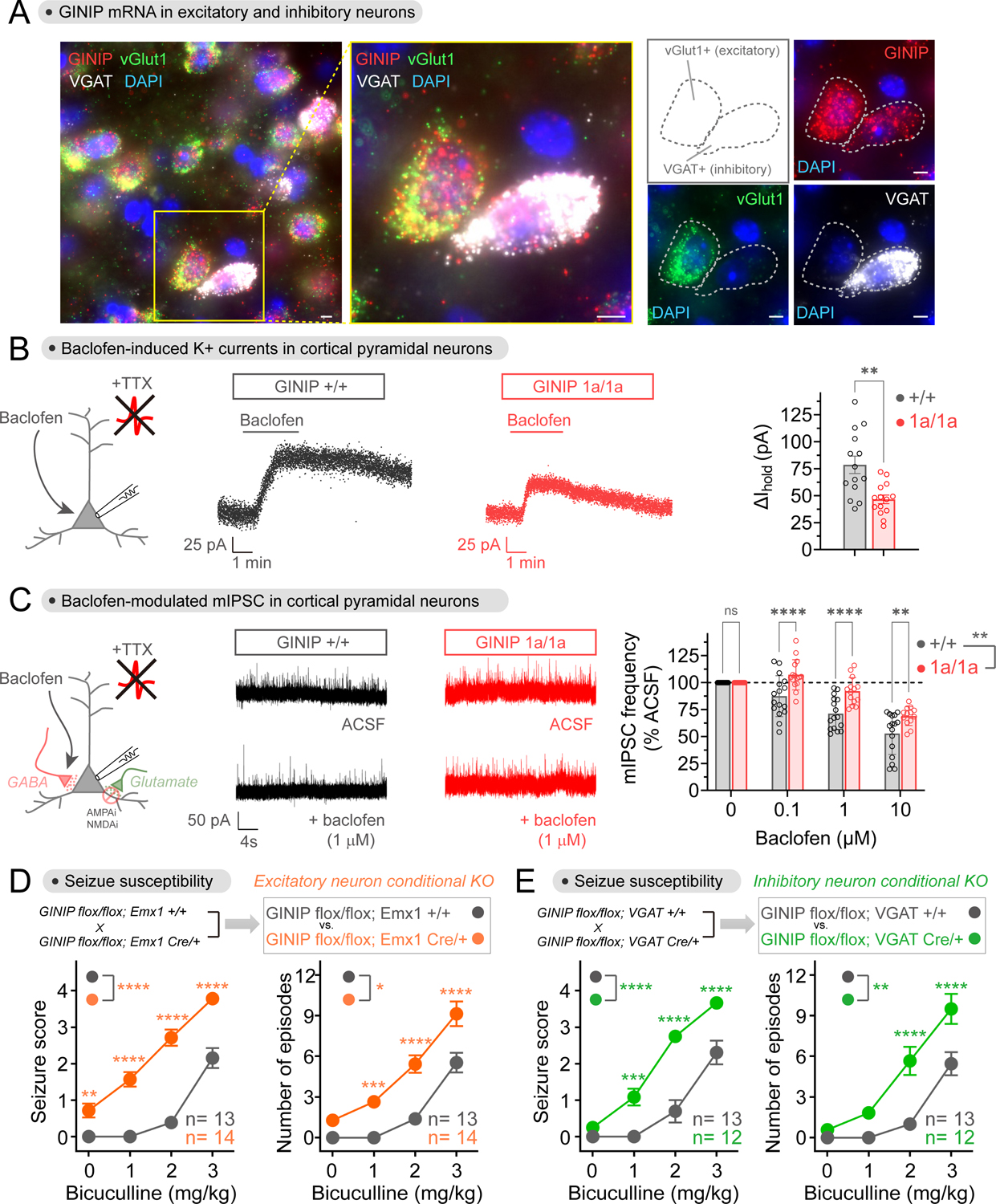
Comment in
-
Built-in functional selectivity in neurons is mediated by the neuronal protein, GINIP.Mol Cell. 2023 Jul 20;83(14):2392-2394. doi: 10.1016/j.molcel.2023.06.034. Mol Cell. 2023. PMID: 37478823
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
