

Ultrasound-responsive low-dose doxorubicin liposomes trigger mitochondrial DNA release and activate cGAS-STING-mediated antitumour immunity
- PMID: 37391428
- PMCID: PMC10313815
- DOI: 10.1038/s41467-023-39607-x
Ultrasound-responsive low-dose doxorubicin liposomes trigger mitochondrial DNA release and activate cGAS-STING-mediated antitumour immunity
Abstract
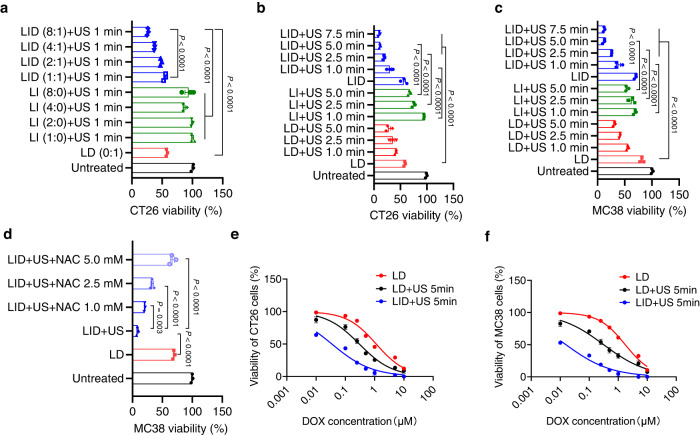
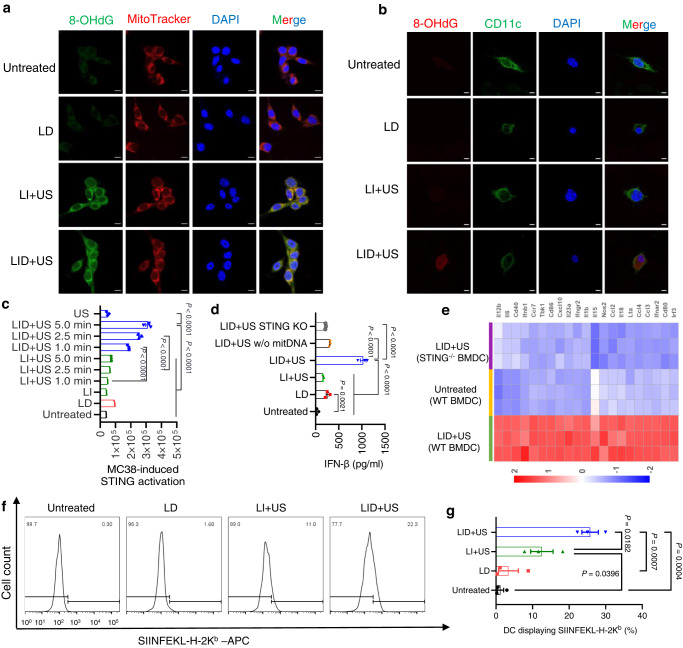
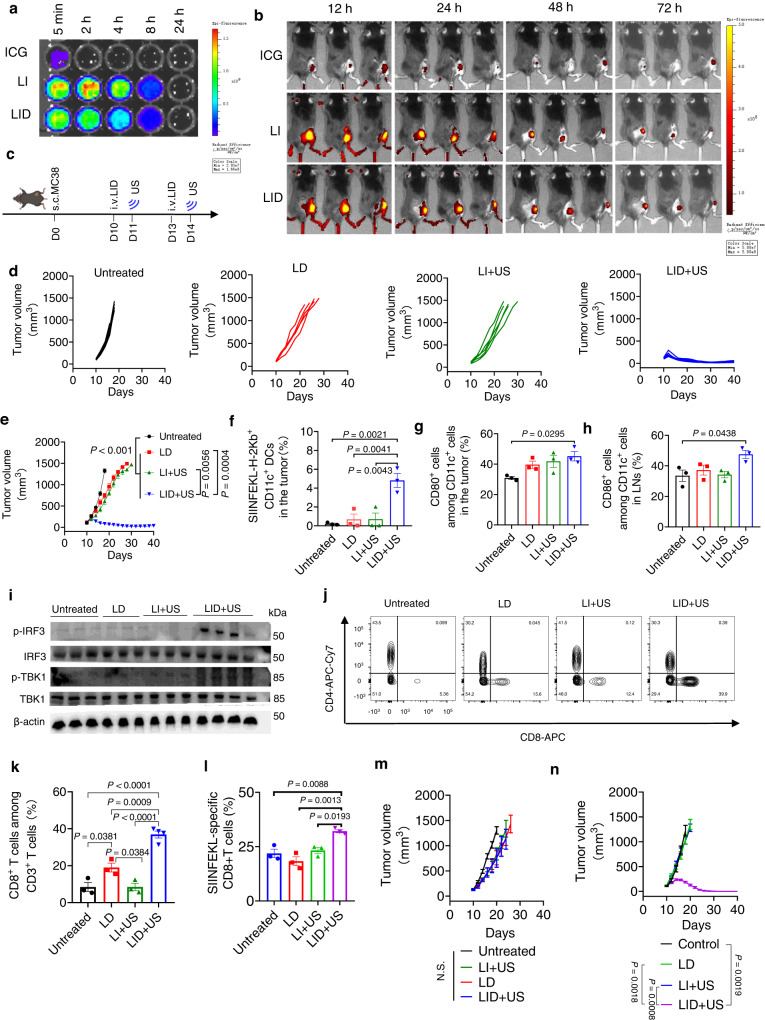
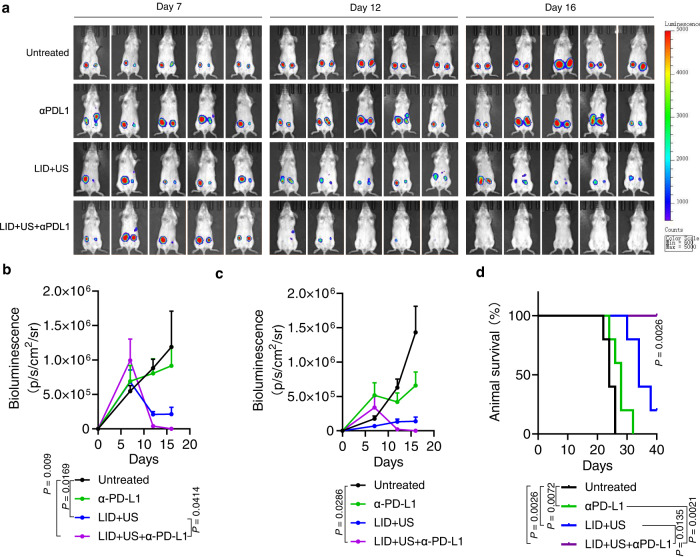
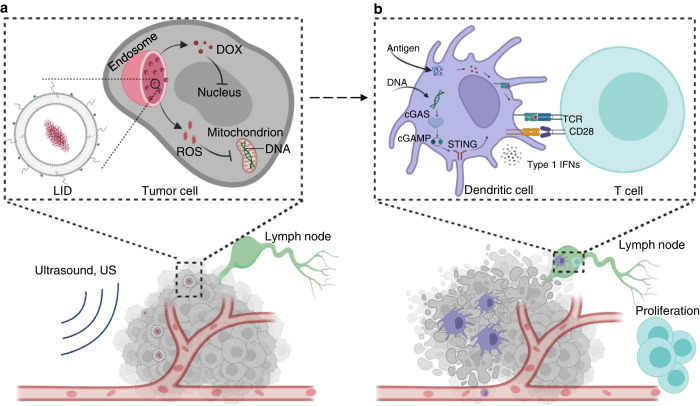
DNA derived from chemotherapeutics-killed tumor cells is one of the most important damage-associated molecular patterns that can activate the cGAS-STING (cyclic GMP-AMP synthase-stimulator of interferon genes) pathway in antigen-presenting cells (APCs) and promote antitumor immunity. However, conventional chemotherapy displays limited tumor cell killing and ineffective transfer of stable tumor DNA to APCs. Here we show that liposomes loaded with an optimized ratio of indocyanine green and doxorubicin, denoted as LID, efficiently generate reactive oxygen species upon exposure to ultrasound. LID plus ultrasound enhance the nuclear delivery of doxorubicin, induce tumor mitochondrial DNA oxidation, and promote oxidized tumor mitochondrial DNA transfer to APCs for effective activation of cGAS-STING signaling. Depleting tumor mitochondrial DNA or knocking out STING in APCs compromises the activation of APCs. Furthermore, systemic injection of LID plus ultrasound over the tumor lead to targeted cytotoxicity and STING activation, eliciting potent antitumor T cell immunity, which upon the combination with immune checkpoint blockade leads to regression of bilateral MC38, CT26, and orthotopic 4T1 tumors in female mice. Our study sheds light on the importance of oxidized tumor mitochondrial DNA in STING-mediated antitumor immunity and may inspire the development of more effective strategies for cancer immunotherapy.
© 2023. The Author(s).
Conflict of interest statement
A patent application (202210306460.1) has been filed based on the ultrasound responsive chemotherapeutics for STING activation, with R.K., C.W., R.Z., and J.H. as inventors. J.M.K. and R.K. hold equity and consult for Corner Therapeutics, a company that has licensed IP generated by J.M.K. and R.K. that may benefit financially if the IP is further validated. J.M.K. has been a paid consultant and or equity holder for multiple biotechnology companies (listed here
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
