

AI in drug discovery and its clinical relevance
- PMID: 37396052
- PMCID: PMC10302550
- DOI: 10.1016/j.heliyon.2023.e17575
AI in drug discovery and its clinical relevance
Abstract
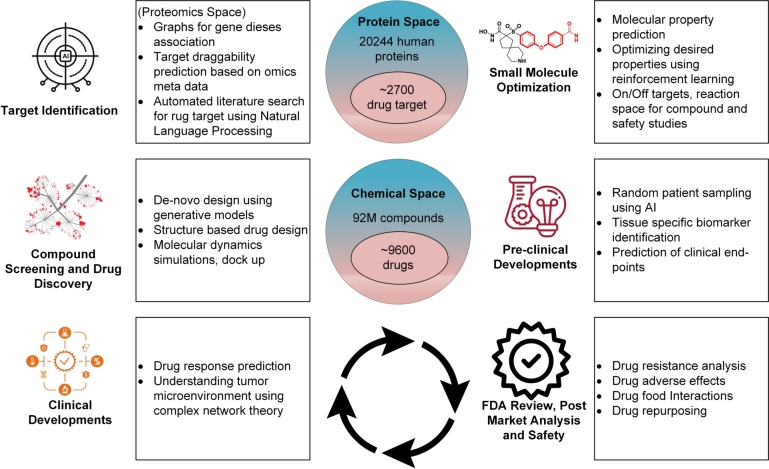
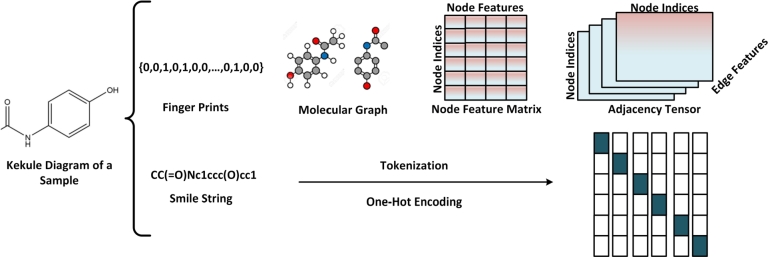
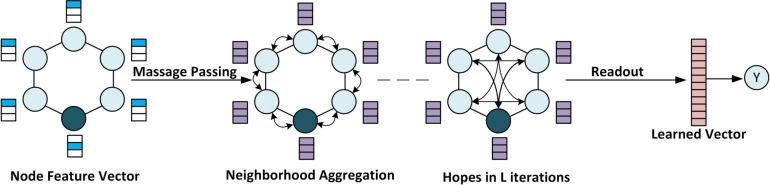
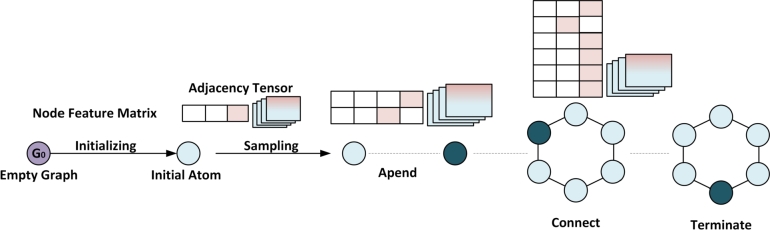
The COVID-19 pandemic has emphasized the need for novel drug discovery process. However, the journey from conceptualizing a drug to its eventual implementation in clinical settings is a long, complex, and expensive process, with many potential points of failure. Over the past decade, a vast growth in medical information has coincided with advances in computational hardware (cloud computing, GPUs, and TPUs) and the rise of deep learning. Medical data generated from large molecular screening profiles, personal health or pathology records, and public health organizations could benefit from analysis by Artificial Intelligence (AI) approaches to speed up and prevent failures in the drug discovery pipeline. We present applications of AI at various stages of drug discovery pipelines, including the inherently computational approaches of de novo design and prediction of a drug's likely properties. Open-source databases and AI-based software tools that facilitate drug design are discussed along with their associated problems of molecule representation, data collection, complexity, labeling, and disparities among labels. How contemporary AI methods, such as graph neural networks, reinforcement learning, and generated models, along with structure-based methods, (i.e., molecular dynamics simulations and molecular docking) can contribute to drug discovery applications and analysis of drug responses is also explored. Finally, recent developments and investments in AI-based start-up companies for biotechnology, drug design and their current progress, hopes and promotions are discussed in this article.
Keywords: Artificial intelligence; Biotechnology; Drug discovery; Graph neural networks; Molecular dynamics simulation; Molecule representation; Reinforcement learning.
© 2023 The Author(s).
Conflict of interest statement
None declared.
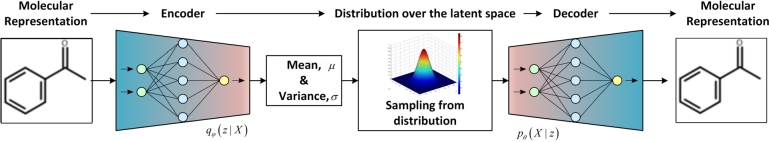
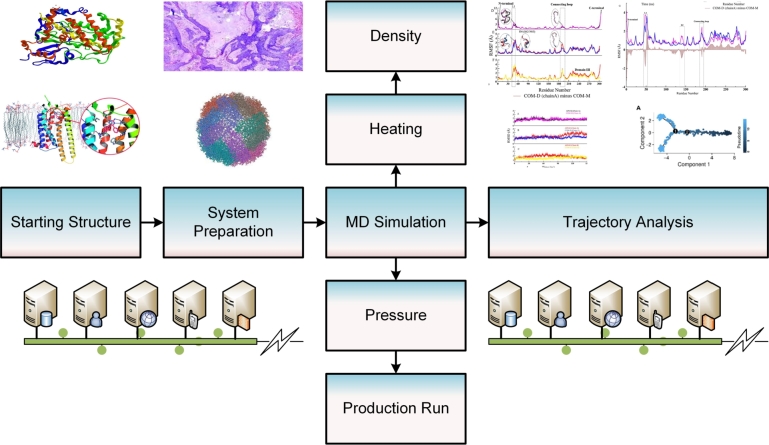
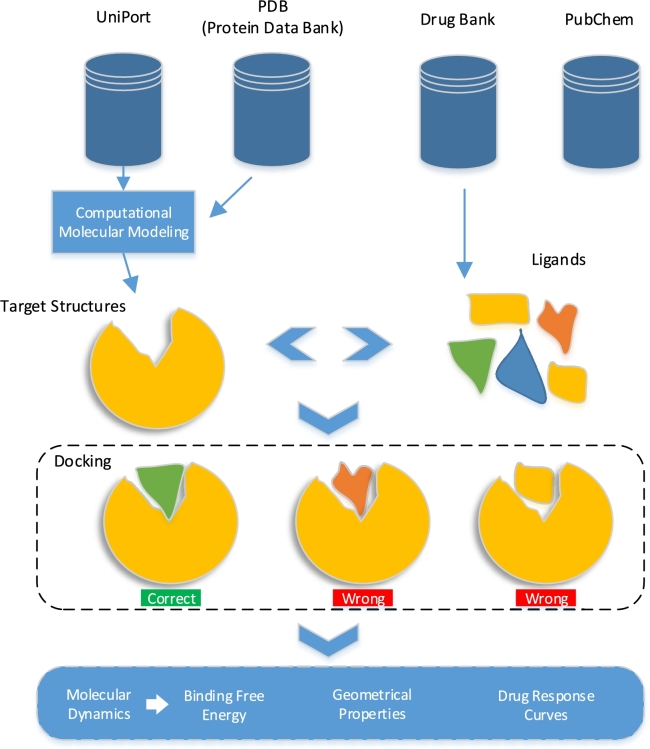
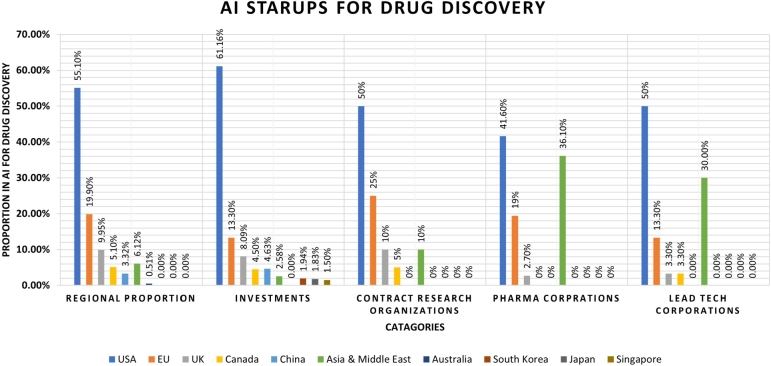
Figures
References
-
- Organization W.H. World Health Organization; 2017. New perspectives on global health spending for universal health coverage. Tech. Rep.
-
- Khanna I. Drug discovery in pharmaceutical industry: productivity challenges and trends. Drug Discov. Today. 2012;17(19–20):1088–1102. - PubMed
-
- Freedman D.H., et al. Hunting for new drugs with ai. Nature. 2019;576(7787):S49–S53. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
