

Immune abnormalities in IgA nephropathy
- PMID: 37398689
- PMCID: PMC10310525
- DOI: 10.1093/ckj/sfad025
Immune abnormalities in IgA nephropathy
Abstract
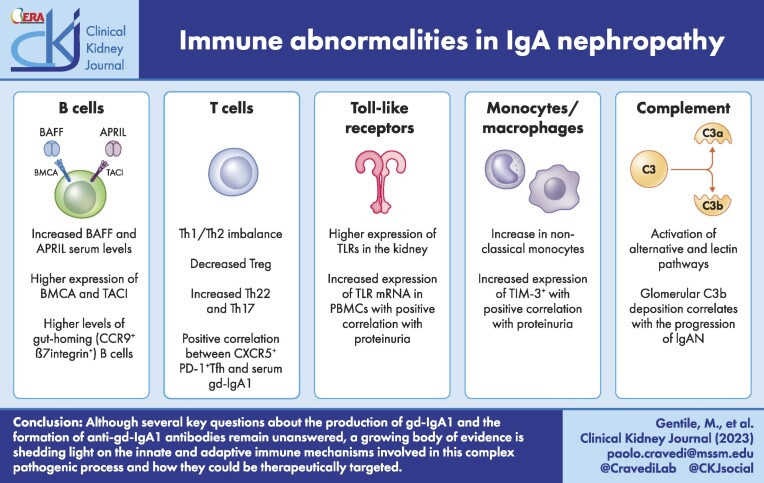
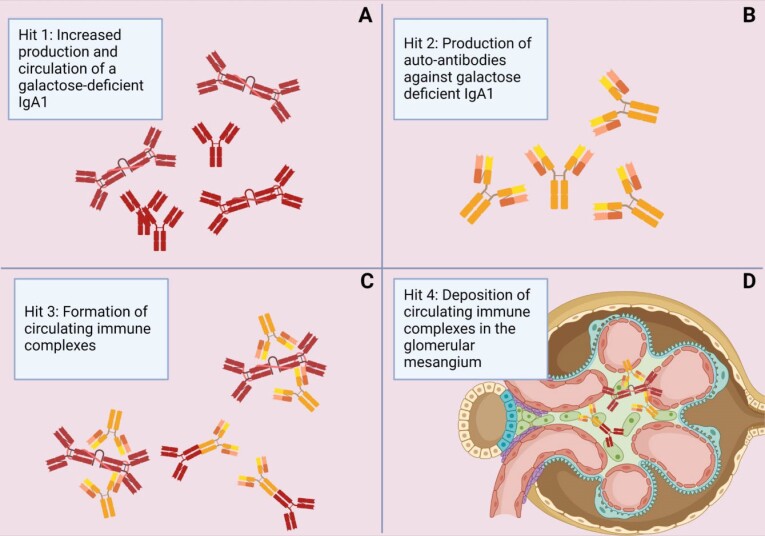
Immunoglobulin A (IgA) nephropathy (IgAN) is the most common primary glomerulonephritis worldwide and it is characterized by mesangial IgA deposition. Asymptomatic hematuria with various degrees of proteinuria is the most common clinical presentation and up to 20%-40% of patients develop end-stage kidney disease within 20 years after disease onset. The pathogenesis of IgAN involves four sequential processes known as the "four-hit hypothesis" which starts with the production of a galactose-deficient IgA1 (gd-IgA1), followed by the formation of anti-gd-IgA1 IgG or IgA1 autoantibodies and immune complexes that ultimately deposit in the glomerular mesangium, leading to inflammation and injury. Although several key questions about the production of gd-IgA1 and the formation of anti-gd-IgA1 antibodies remain unanswered, a growing body of evidence is shedding light on the innate and adaptive immune mechanisms involved in this complex pathogenic process. Herein, we will focus on these mechanisms that, along with genetic and environmental factors, are thought to play a key role in disease pathogenesis.
Keywords: IgA nephropathy; adaptive immunity; galactose-deficient IgA1; innate immunity.
© The Author(s) 2023. Published by Oxford University Press on behalf of the ERA.
Conflict of interest statement
Paolo Cravedi is an advisor for Chinook Therapeutics and Calliditas Therapeutics. The other authors have no conflict of interest to declare.
Figures

References
-
- Berger J. IgA glomerular deposits in renal disease. Transpl Proc 1969;1:939–44. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
