

SMARCA4 mutation causes human otosclerosis and a similar phenotype in mice
- PMID: 37399313
- PMCID: PMC10756932
- DOI: 10.1136/jmg-2023-109264
SMARCA4 mutation causes human otosclerosis and a similar phenotype in mice
Abstract
Background: Otosclerosis is a common cause of adult-onset progressive hearing loss, affecting 0.3%-0.4% of the population. It results from dysregulation of bone homeostasis in the otic capsule, most commonly leading to fixation of the stapes bone, impairing sound conduction through the middle ear. Otosclerosis has a well-known genetic predisposition including familial cases with apparent autosomal dominant mode of inheritance. While linkage analysis and genome-wide association studies suggested an association with several genomic loci and with genes encoding structural proteins involved in bone formation or metabolism, the molecular genetic pathophysiology of human otosclerosis is yet mostly unknown.
Methods: Whole-exome sequencing, linkage analysis, generation of CRISPR mutant mice, hearing tests and micro-CT.
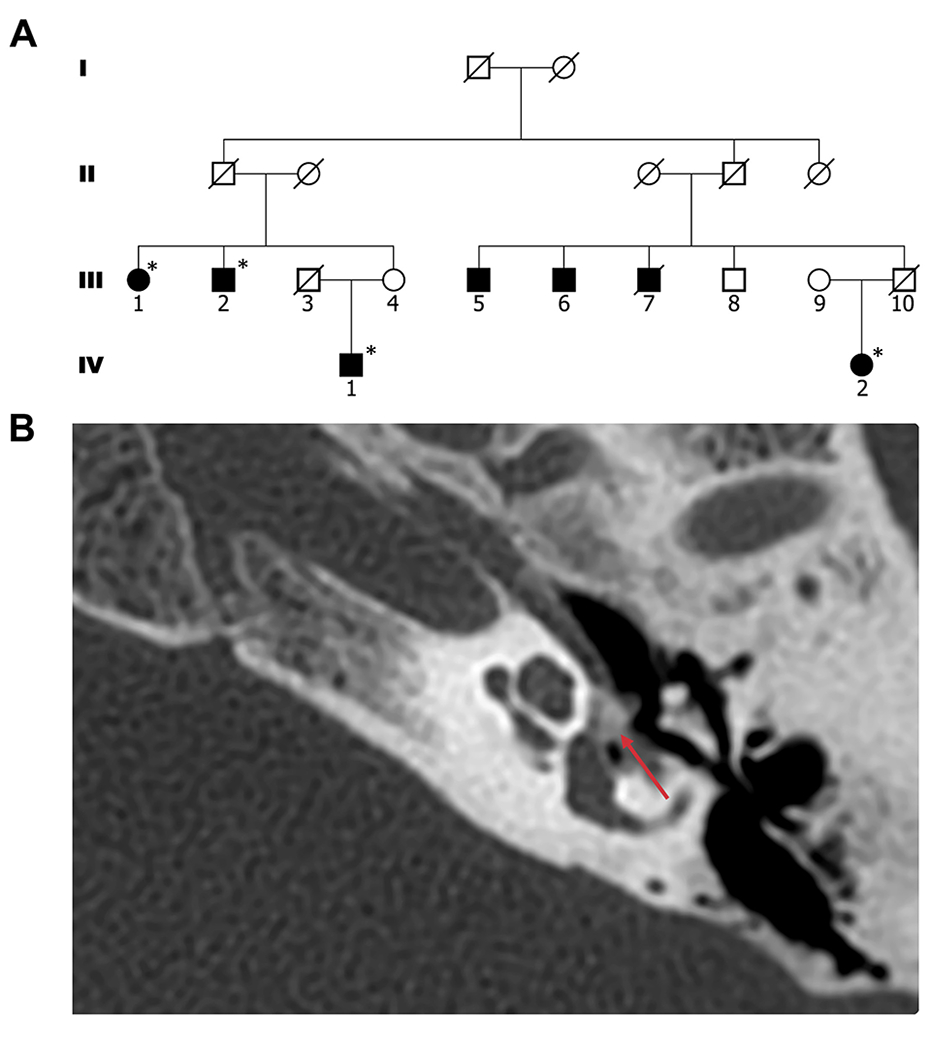
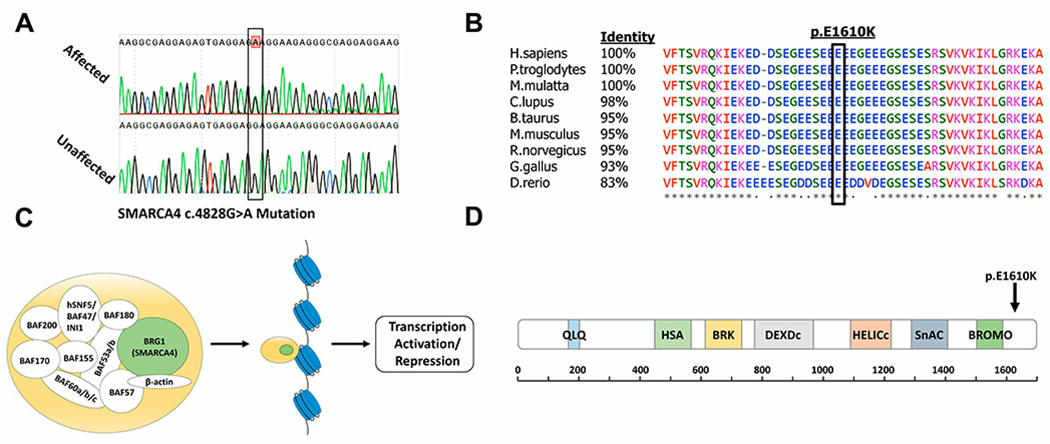
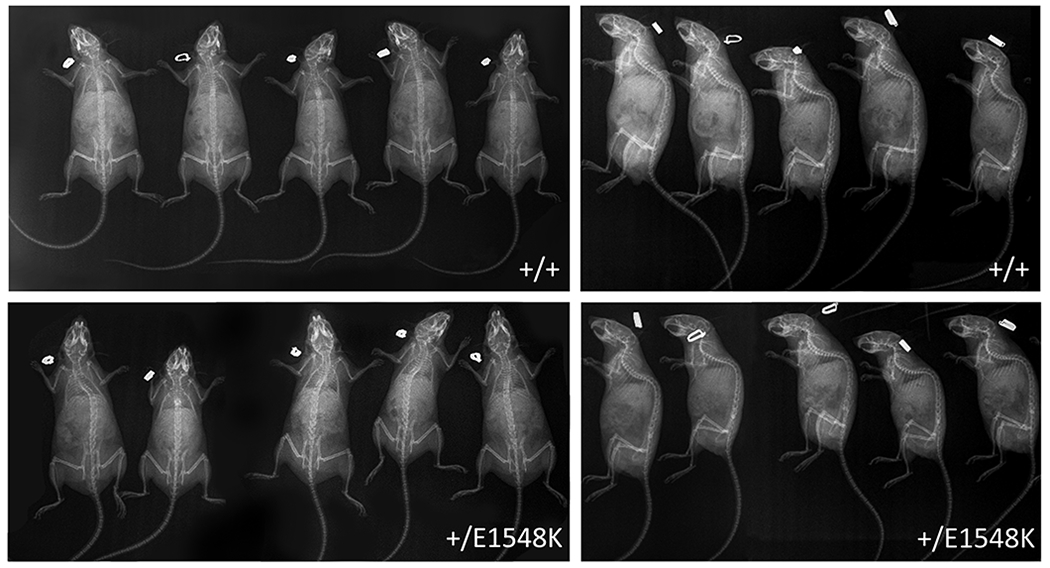
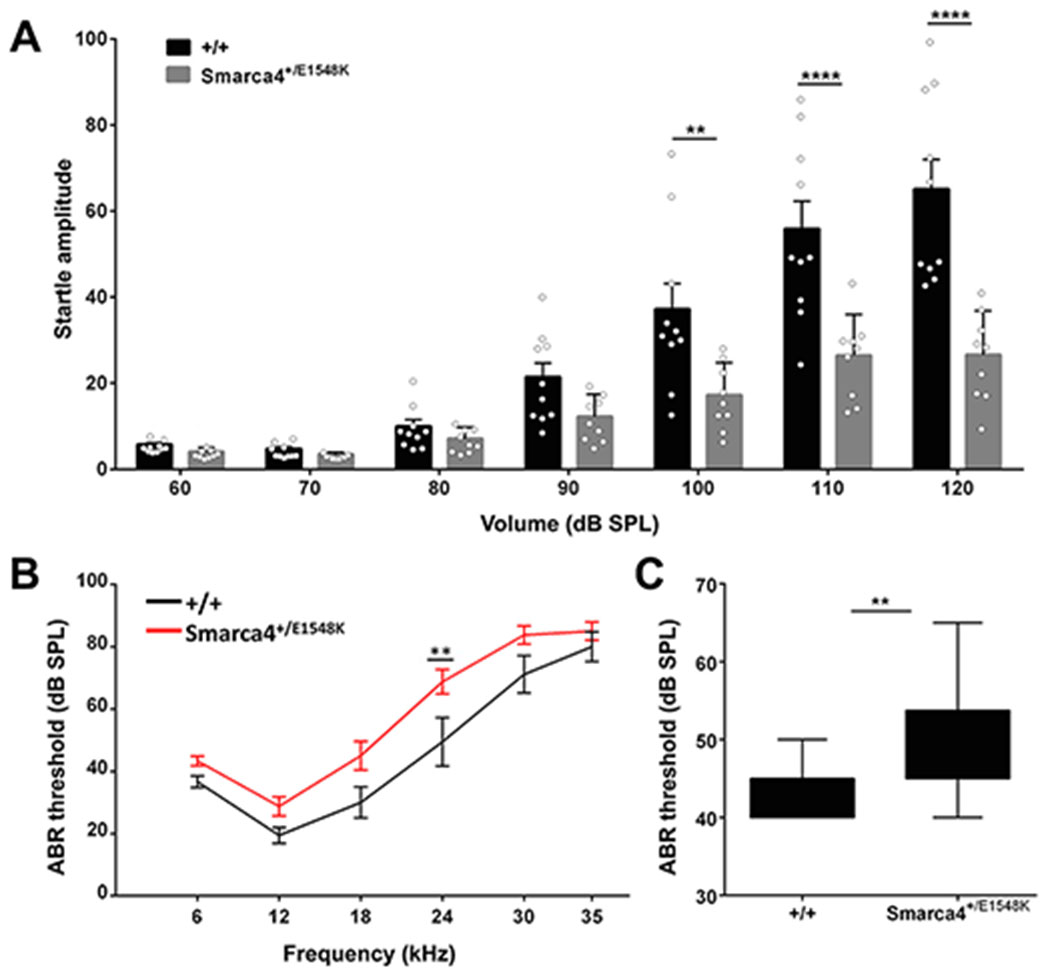
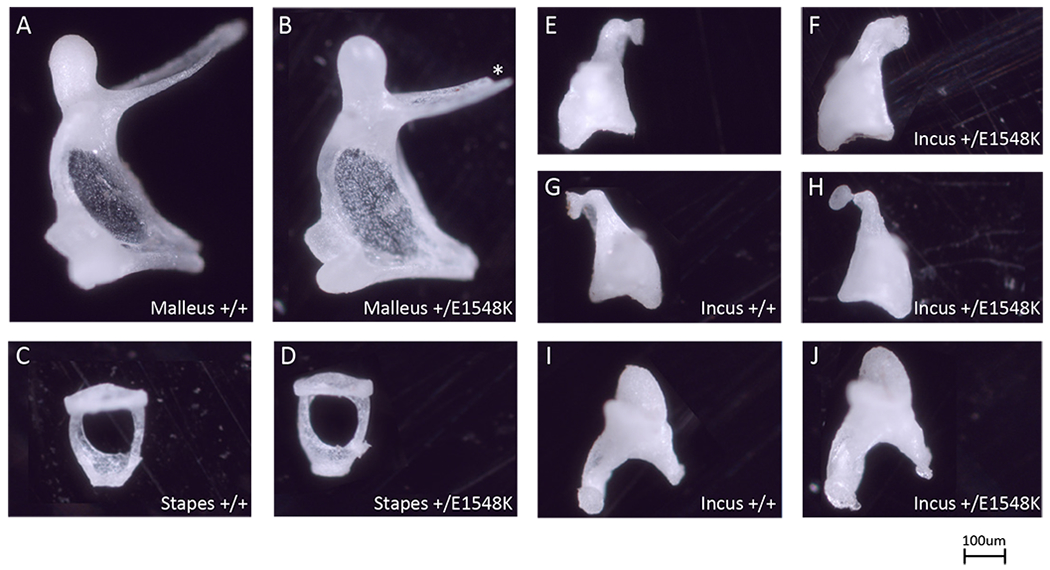
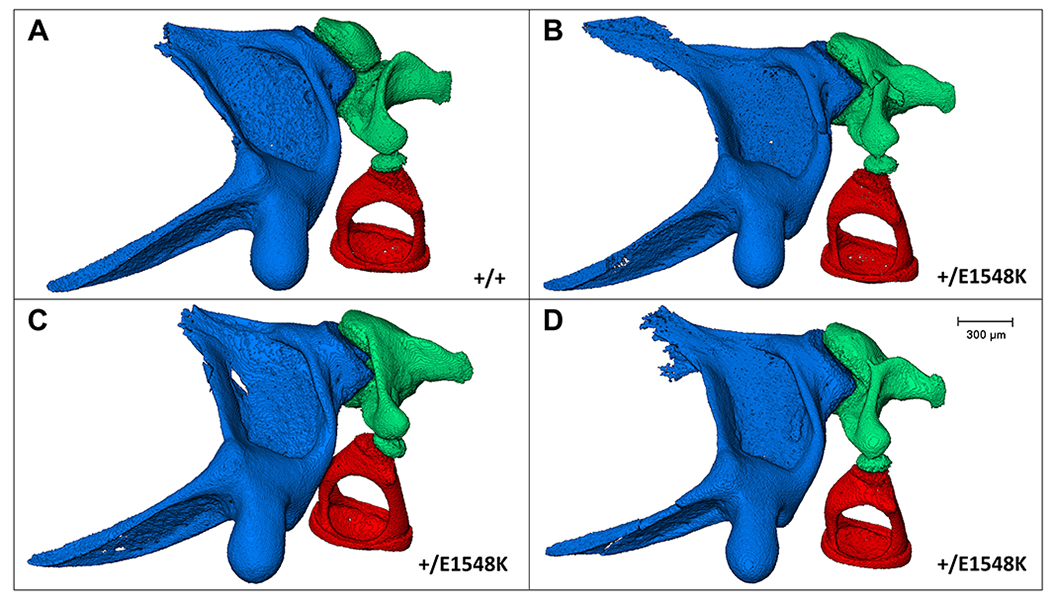
Results: Through genetic studies of kindred with seven individuals affected by apparent autosomal dominant otosclerosis, we identified a disease-causing variant in SMARCA4, encoding a key component of the PBAF chromatin remodelling complex. We generated CRISPR-Cas9 transgenic mice carrying the human mutation in the mouse SMARCA4 orthologue. Mutant Smarca4+/E1548K mice exhibited marked hearing impairment demonstrated through acoustic startle response and auditory brainstem response tests. Isolated ossicles of the auditory bullae of mutant mice exhibited a highly irregular structure of the incus bone, and their in situ micro-CT studies demonstrated the anomalous structure of the incus bone, causing disruption in the ossicular chain.
Conclusion: We demonstrate that otosclerosis can be caused by a variant in SMARCA4, with a similar phenotype of hearing impairment and abnormal bone formation in the auditory bullae in transgenic mice carrying the human mutation in the mouse SMARCA4 orthologue.
Keywords: genetics; human genetics; molecular biology; mutation, missense; phenotype.
© Author(s) (or their employer(s)) 2024. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Declau F, Van Spaendonck M, Timmermans JP, et al. Prevalence of otosclerosis in an unselected series of temporal bones. Otol Neurotol 2001;22:596–602. - PubMed
-
- Gordon MA. The genetics of otosclerosis: A review. Otol Neurotol 1989;10:426–38. - PubMed
-
- Quesnel AM, Ishai R, McKenna MJ. Otosclerosis: Temporal Bone Pathology. Otolaryngol Clin North Am 2018;51:291–303. - PubMed
-
- Chole RA, McKenna M. Pathophysiology of Otosclerosis. Otol Neurotol 2001;22:249–57. - PubMed
-
- Rudic M, Keogh I, Wagner R, et al. The pathophysiology of otosclerosis: Review of current research. Hear Res 2015;330:51–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous