

Comprehensive tissue deconvolution of cell-free DNA by deep learning for disease diagnosis and monitoring
- PMID: 37399400
- PMCID: PMC10334733
- DOI: 10.1073/pnas.2305236120
Comprehensive tissue deconvolution of cell-free DNA by deep learning for disease diagnosis and monitoring
Erratum in
-
Correction for Li et al., Comprehensive tissue deconvolution of cell-free DNA by deep learning for disease diagnosis and monitoring.Proc Natl Acad Sci U S A. 2025 Oct 7;122(40):e2525325122. doi: 10.1073/pnas.2525325122. Epub 2025 Oct 1. Proc Natl Acad Sci U S A. 2025. PMID: 41032527 Free PMC article. No abstract available.
Abstract
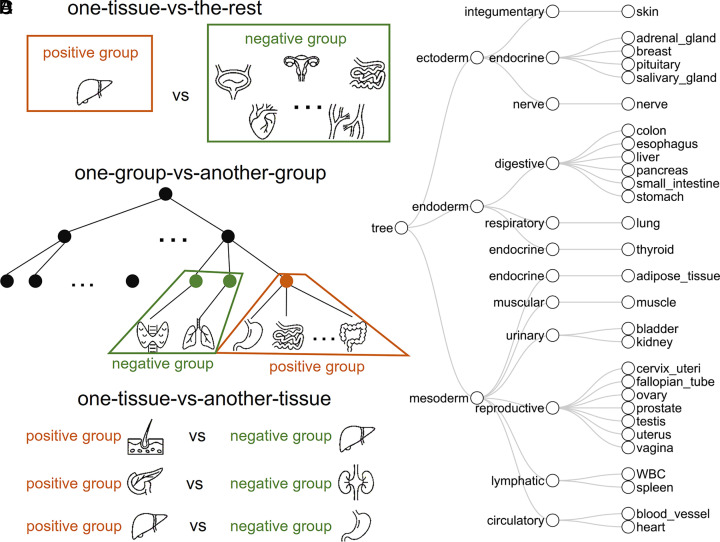
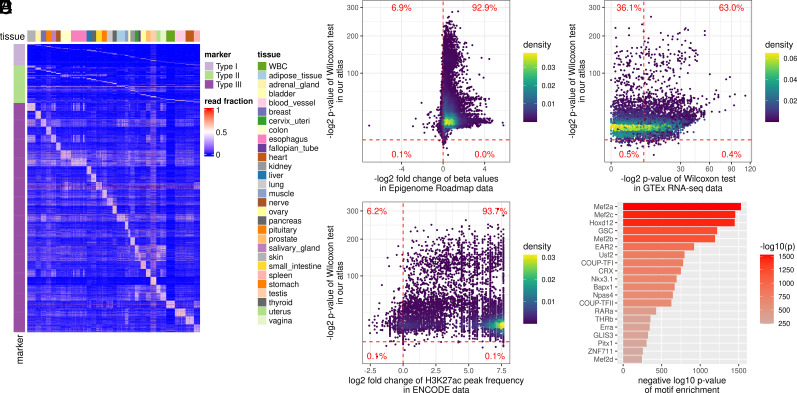
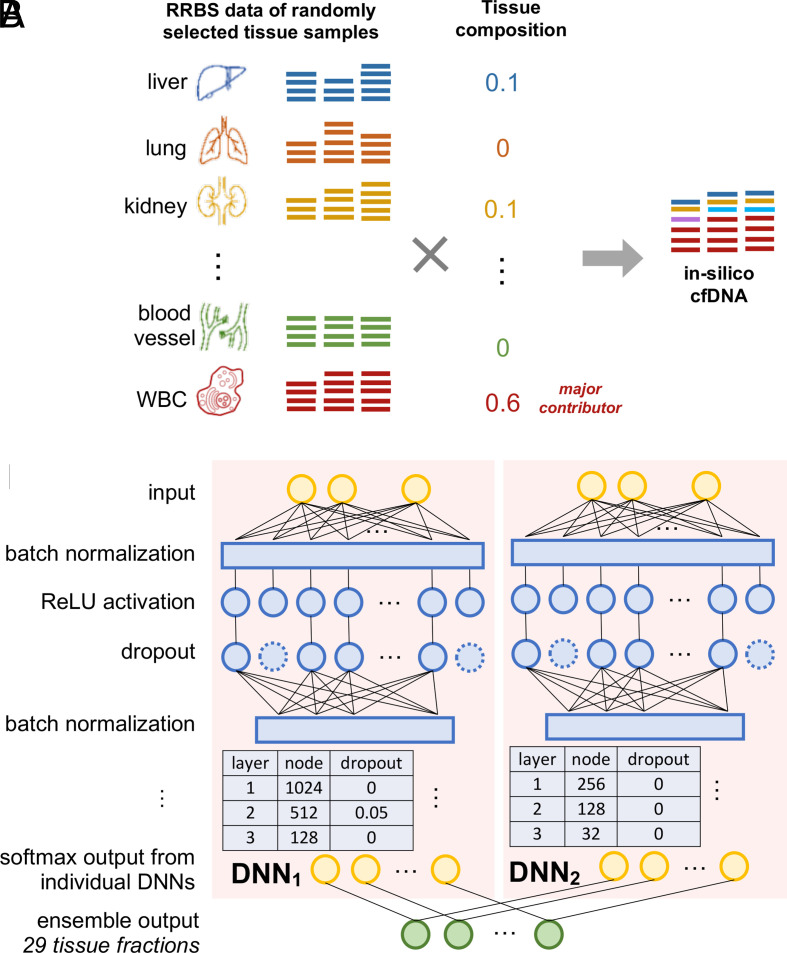
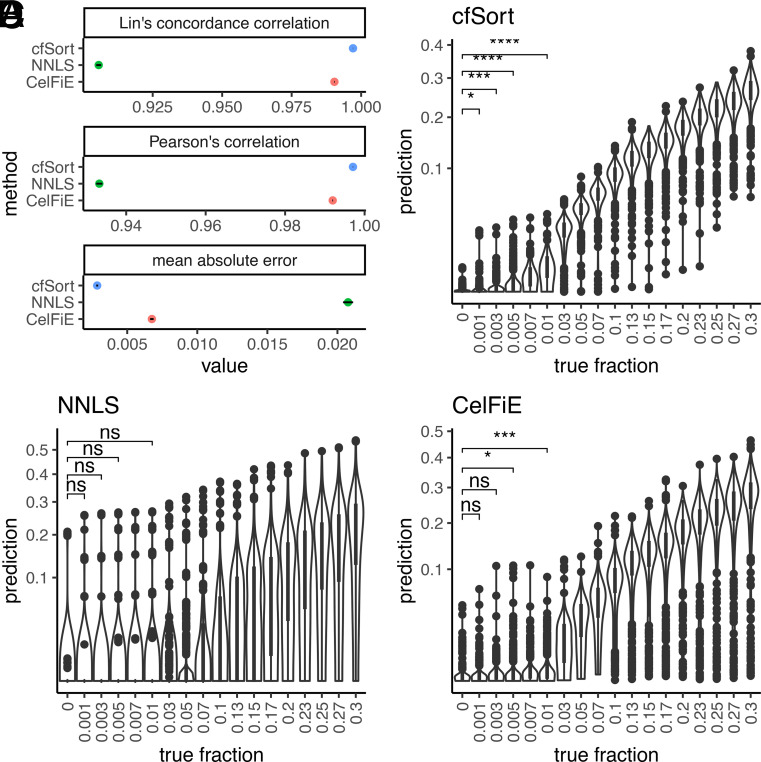
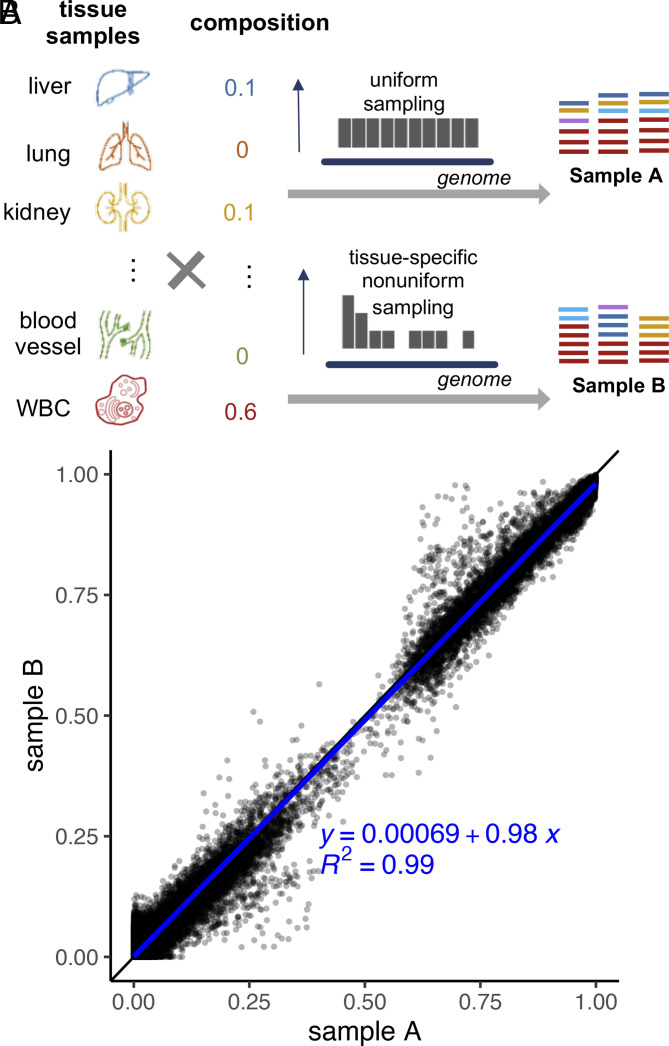
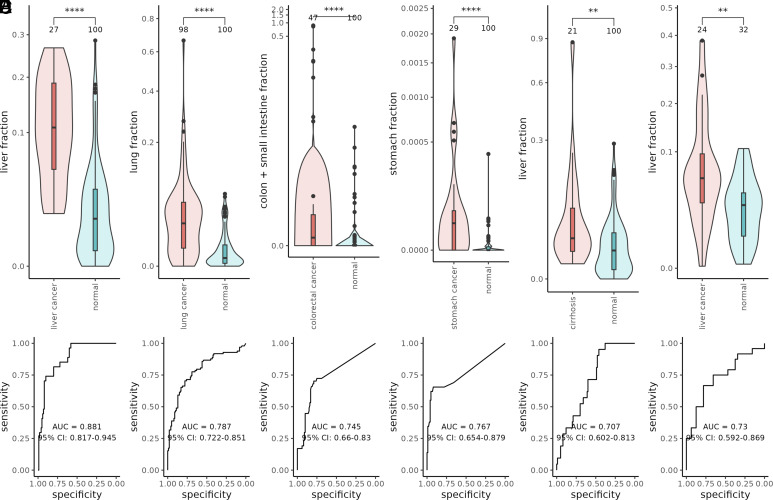
Plasma cell-free DNA (cfDNA) is a noninvasive biomarker for cell death of all organs. Deciphering the tissue origin of cfDNA can reveal abnormal cell death because of diseases, which has great clinical potential in disease detection and monitoring. Despite the great promise, the sensitive and accurate quantification of tissue-derived cfDNA remains challenging to existing methods due to the limited characterization of tissue methylation and the reliance on unsupervised methods. To fully exploit the clinical potential of tissue-derived cfDNA, here we present one of the largest comprehensive and high-resolution methylation atlas based on 521 noncancer tissue samples spanning 29 major types of human tissues. We systematically identified fragment-level tissue-specific methylation patterns and extensively validated them in orthogonal datasets. Based on the rich tissue methylation atlas, we develop the first supervised tissue deconvolution approach, a deep-learning-powered model, cfSort, for sensitive and accurate tissue deconvolution in cfDNA. On the benchmarking data, cfSort showed superior sensitivity and accuracy compared to the existing methods. We further demonstrated the clinical utilities of cfSort with two potential applications: aiding disease diagnosis and monitoring treatment side effects. The tissue-derived cfDNA fraction estimated from cfSort reflected the clinical outcomes of the patients. In summary, the tissue methylation atlas and cfSort enhanced the performance of tissue deconvolution in cfDNA, thus facilitating cfDNA-based disease detection and longitudinal treatment monitoring.
Keywords: DNA methylation; cell-free DNA; disease diagnosis; disease monitoring; tissue deconvolution.
Conflict of interest statement
W.L., W.H.W., and X.J.Z. are co-founders of EarlyDiagnostics Inc. X.N. and M.L.S. are employees at EarlyDiagnostics Inc. S.L. is a former employee at EarlyDiagnostics Inc. S.L., W.Z., X.N., W.L., M.L.S., Y.Z., W.H.W., S.M.D., and X.J.Z. own stocks of EarlyDiagnostics Inc. The other authors declare no competing interests.
Figures

References
-
- Wan J., et al. , Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 17, 223–238 (2017). - PubMed
-
- Stroun M., et al. , The origin and mechanism of circulating DNA. Ann. N. Y. Acad. Sci. 906, 161–168 (2000). - PubMed
-
- Li S., et al. , cfTrack: A method of exome-wide mutation analysis of cell-free DNA to simultaneously monitor the full spectrum of cancer treatment outcomes including MRD, recurrence, and evolutioncfTrack: Comprehensive cancer monitoring using cfDNA. Clin. Cancer Res. 28, 1841–1853 (2022). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
