

Exploring switch II pocket conformation of KRAS(G12D) with mutant-selective monobody inhibitors
- PMID: 37399416
- PMCID: PMC10334749
- DOI: 10.1073/pnas.2302485120
Exploring switch II pocket conformation of KRAS(G12D) with mutant-selective monobody inhibitors
Abstract
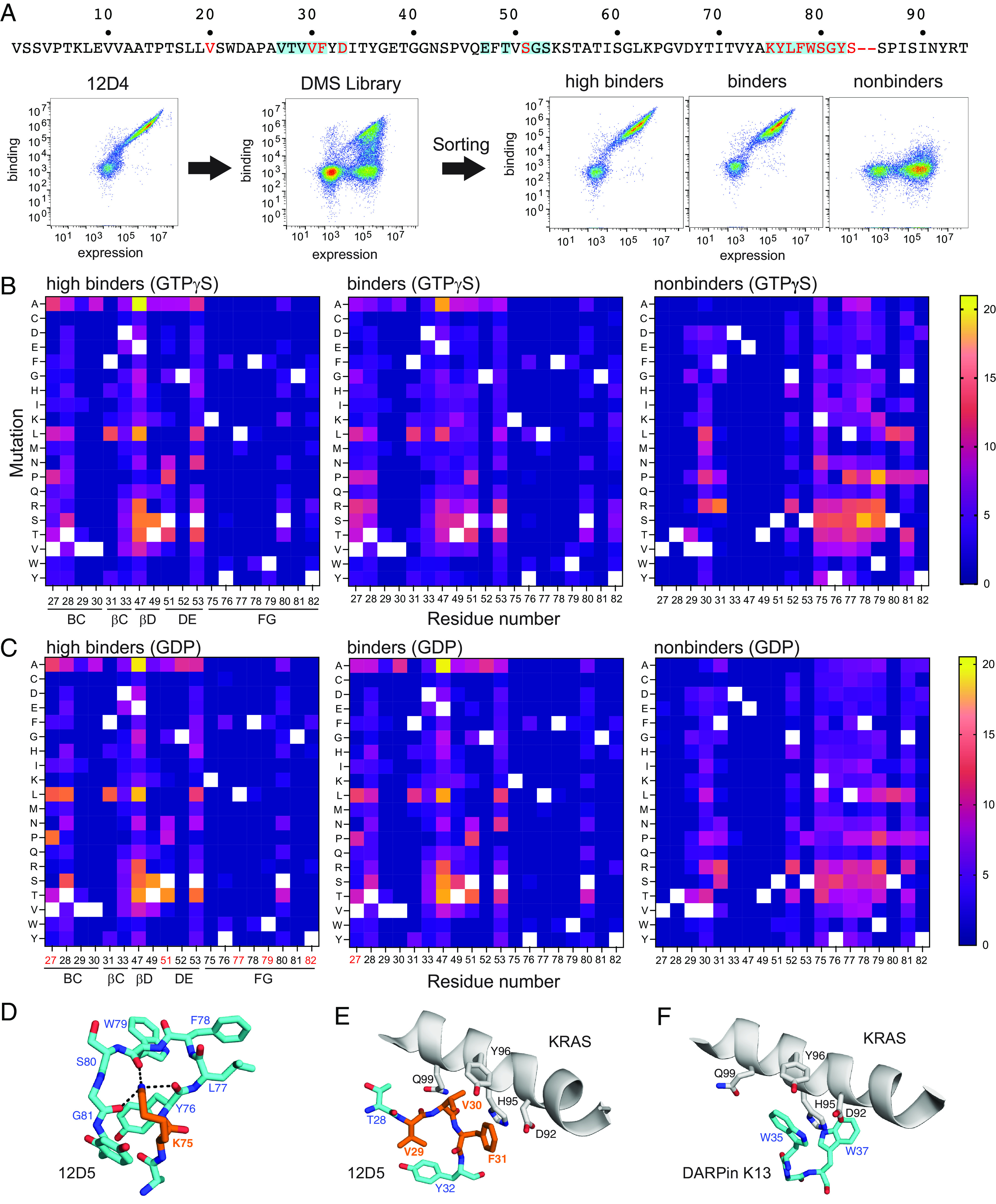
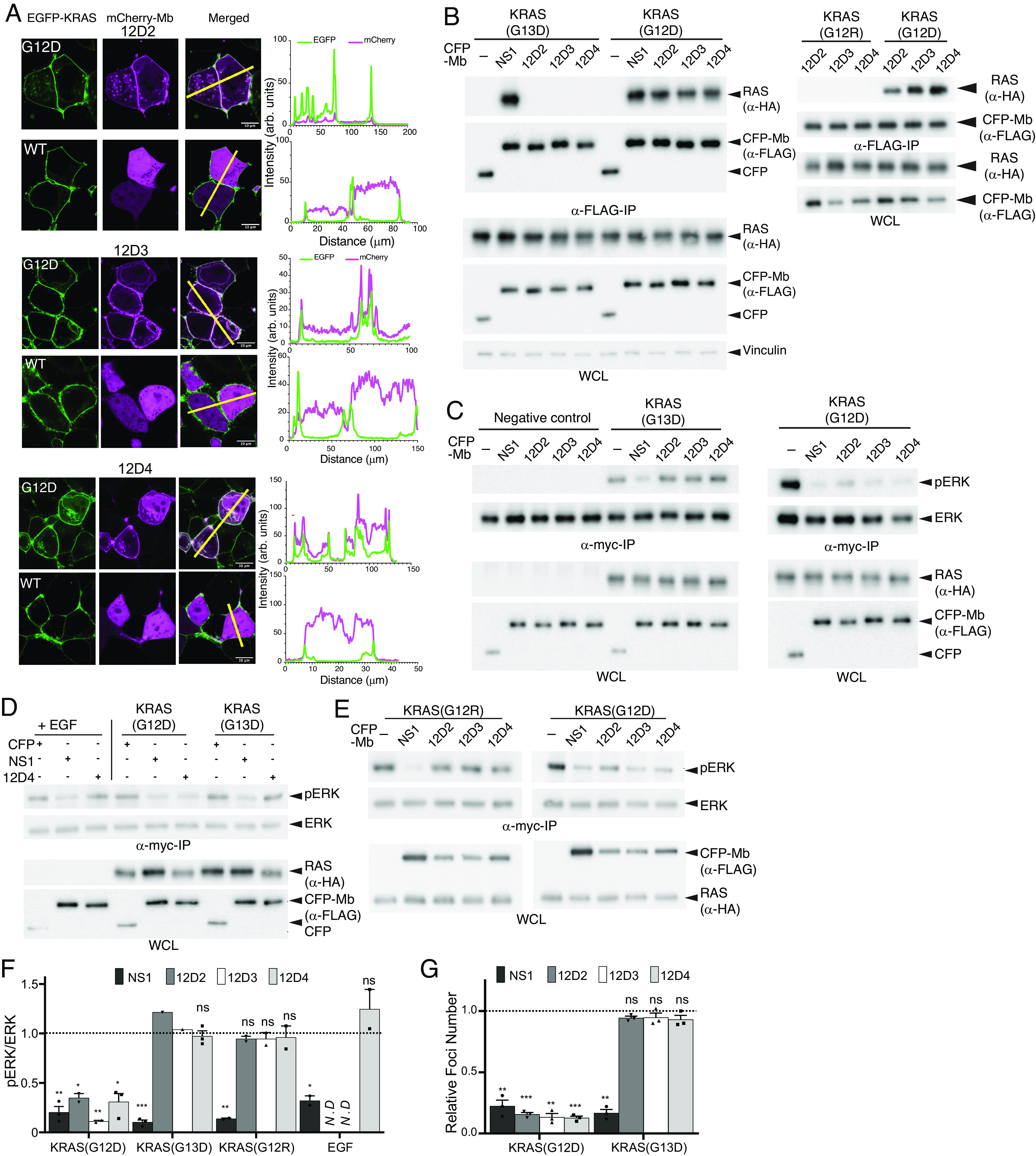
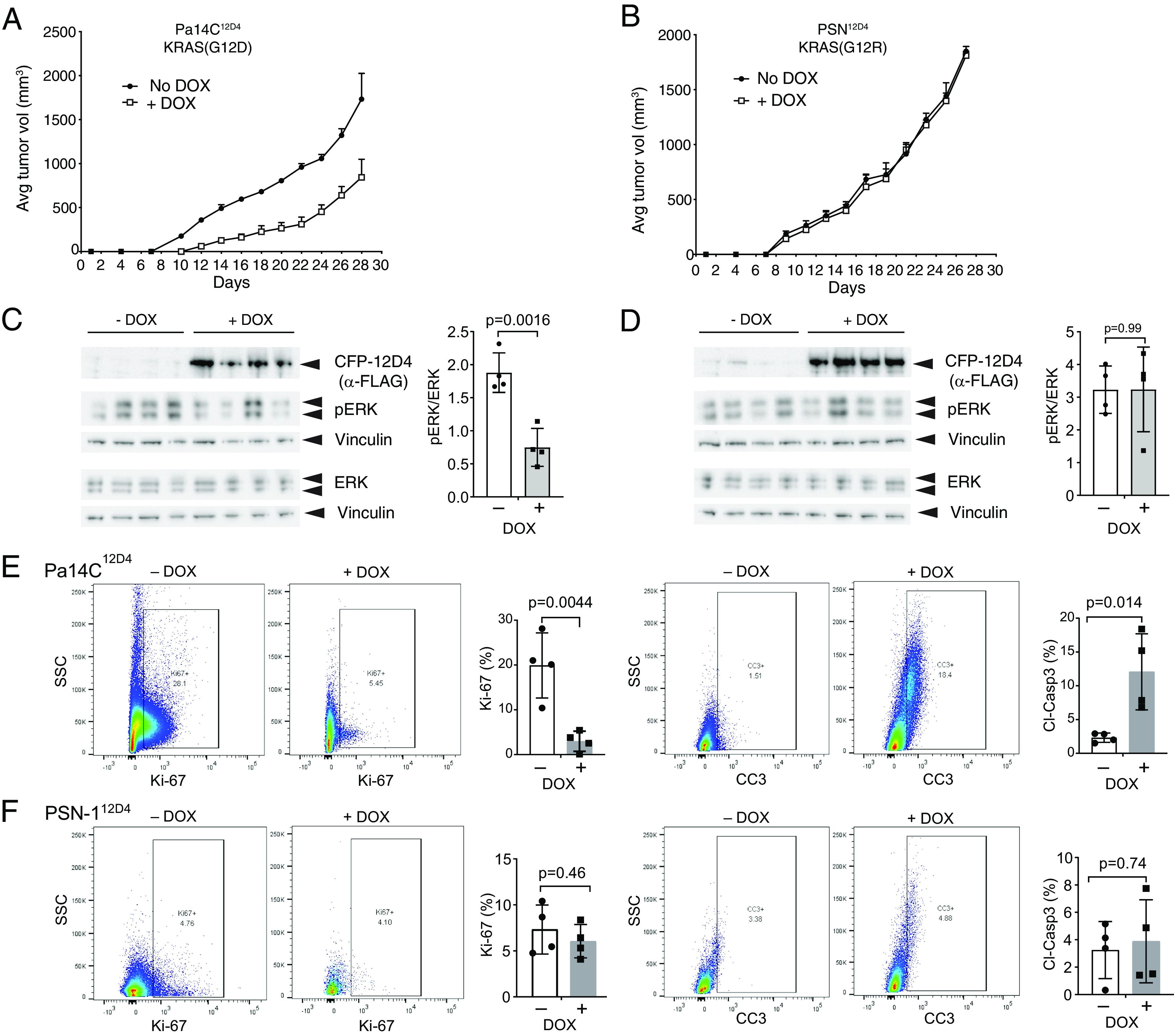
The G12D mutation is among the most common KRAS mutations associated with cancer, in particular, pancreatic cancer. Here, we have developed monobodies, small synthetic binding proteins, that are selective to KRAS(G12D) over KRAS(wild type) and other oncogenic KRAS mutations, as well as over the G12D mutation in HRAS and NRAS. Crystallographic studies revealed that, similar to other KRAS mutant-selective inhibitors, the initial monobody bound to the S-II pocket, the groove between switch II and α3 helix, and captured this pocket in the most widely open form reported to date. Unlike other G12D-selective polypeptides reported to date, the monobody used its backbone NH group to directly recognize the side chain of KRAS Asp12, a feature that closely resembles that of a small-molecule inhibitor, MTRX1133. The monobody also directly interacted with H95, a residue not conserved in RAS isoforms. These features rationalize the high selectivity toward the G12D mutant and the KRAS isoform. Structure-guided affinity maturation resulted in monobodies with low nM KD values. Deep mutational scanning of a monobody generated hundreds of functional and nonfunctional single-point mutants, which identified crucial residues for binding and those that contributed to the selectivity toward the GTP- and GDP-bound states. When expressed in cells as genetically encoded reagents, these monobodies engaged selectively with KRAS(G12D) and inhibited KRAS(G12D)-mediated signaling and tumorigenesis. These results further illustrate the plasticity of the S-II pocket, which may be exploited for the design of next-generation KRAS(G12D)-selective inhibitors.
Keywords: conformational plasticity; drug discovery; intracellular biologics; protein engineering; protein-protein interaction.
Conflict of interest statement
S.K. is a co-founder and holds equity in Aethon Therapeutics and Revalia Bio. P.A., T.H., A.K., K.W.T., and S.K. are listed as inventors of pending patents on technology described in this paper filed by New York University. A.K. and S.K. are listed as inventors on issued and pending patents on the monobody technology filed by the University of Chicago and Novartis. S.K. has received research funding from Aethon Therapeutics, Argenx BVBA, Black Diamond Therapeutics, and Puretech Health. S.K. received consulting fees from Aethon Therapeutics and Black Diamond Therapeutics.
Figures
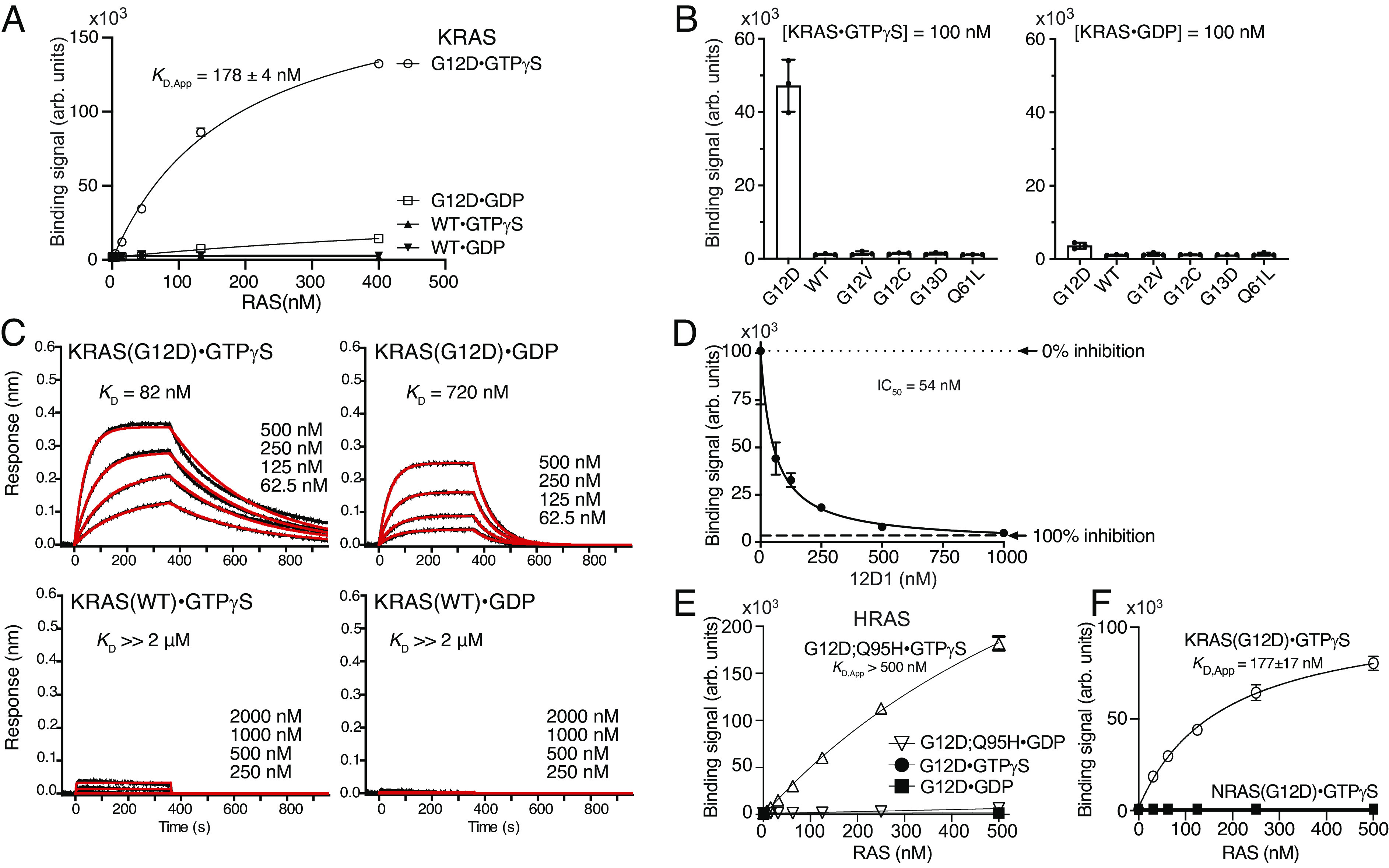
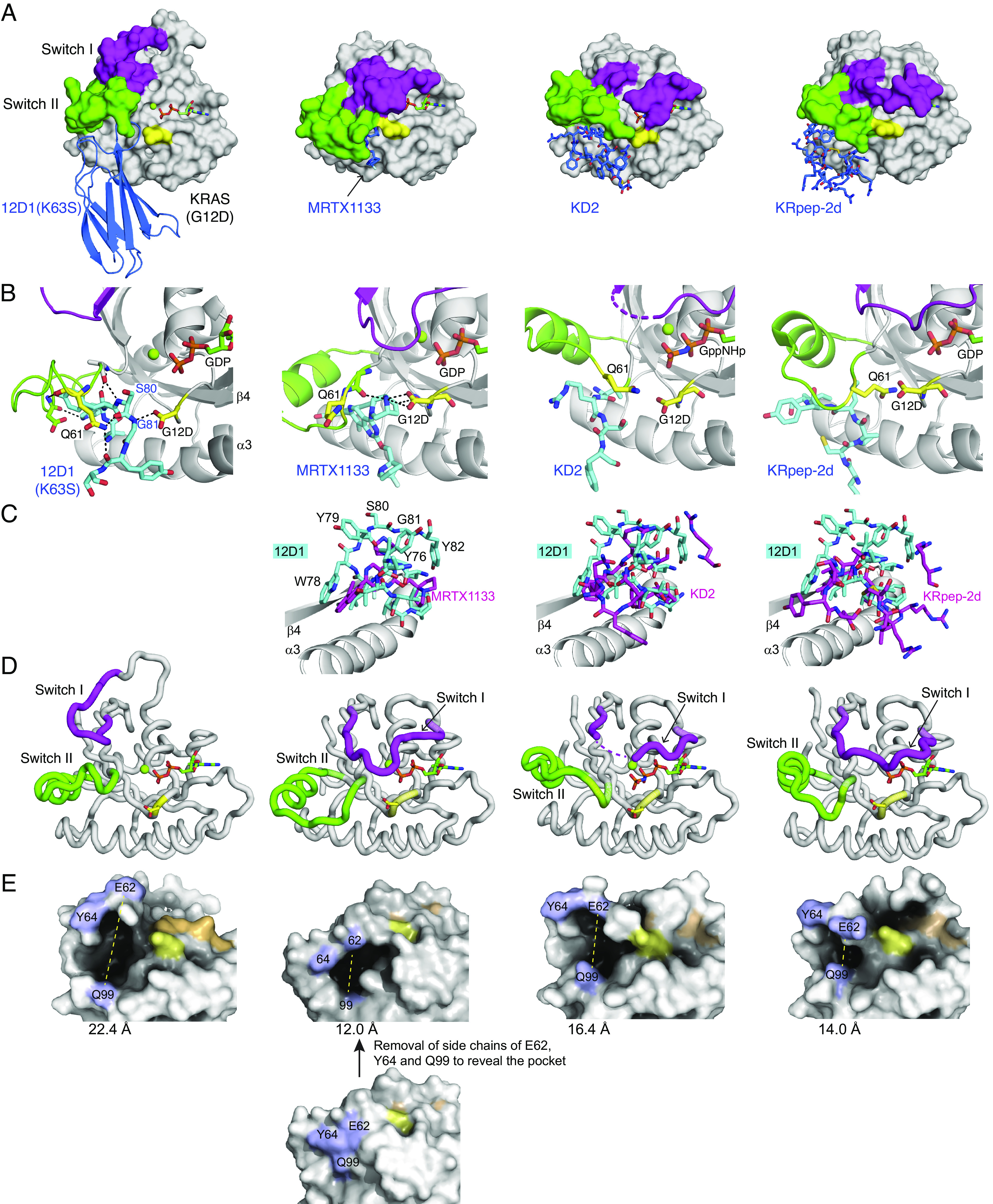
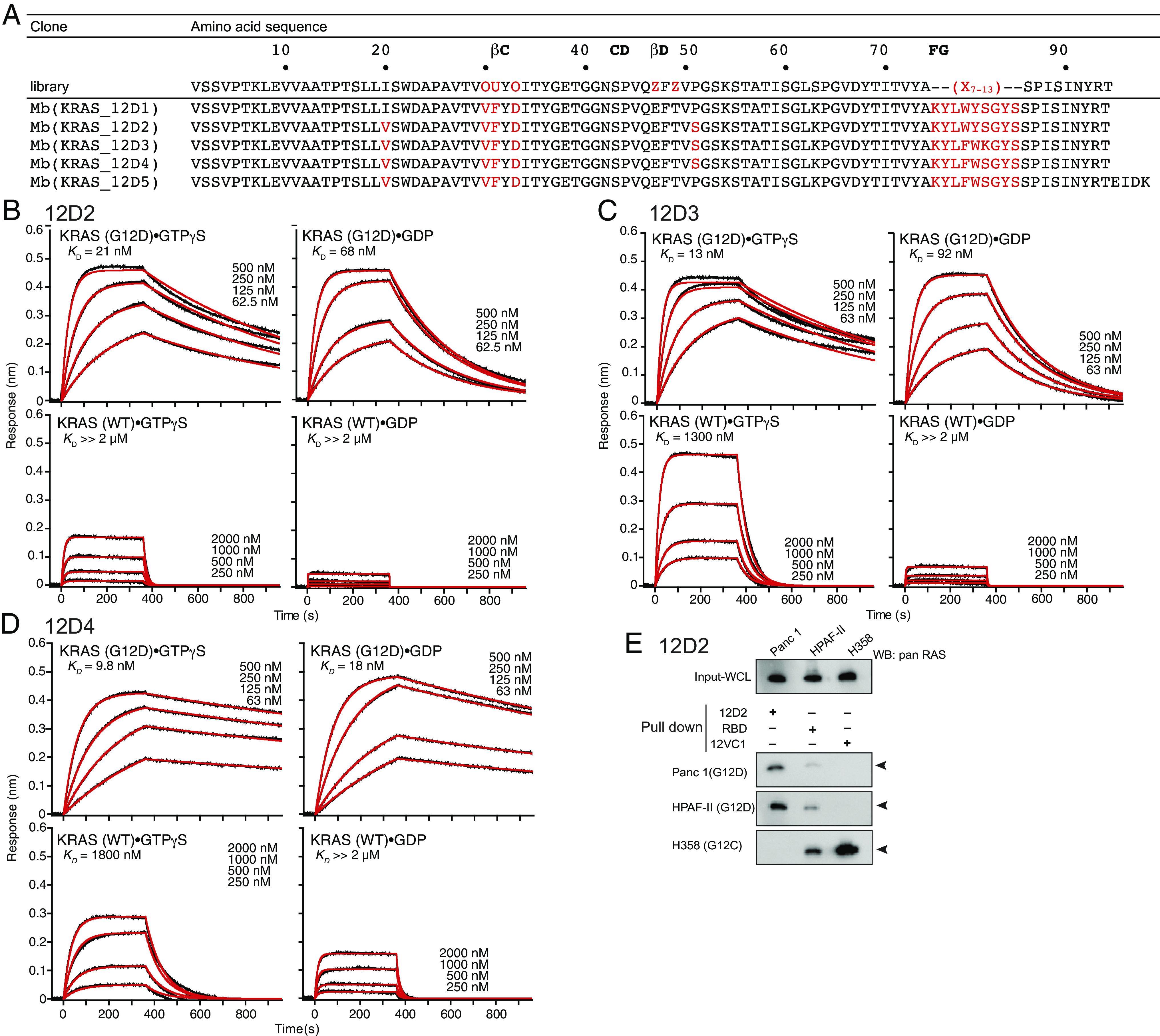
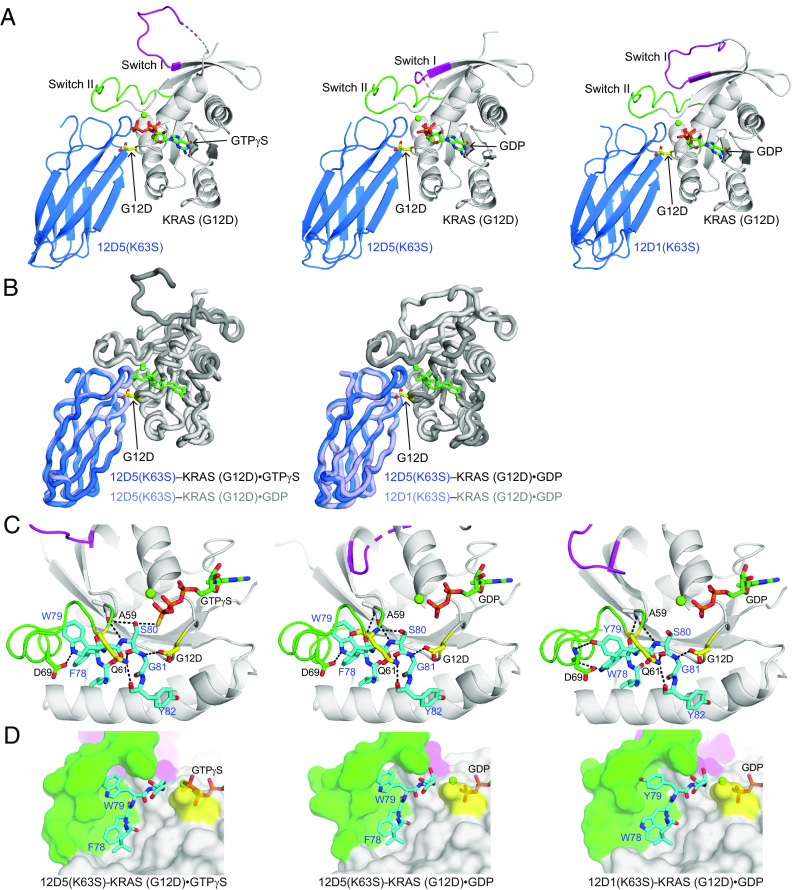
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
