

Extrasynaptic NMDA receptors in acute and chronic excitotoxicity: implications for preventive treatments of ischemic stroke and late-onset Alzheimer's disease
- PMID: 37400870
- PMCID: PMC10318843
- DOI: 10.1186/s13024-023-00636-1
Extrasynaptic NMDA receptors in acute and chronic excitotoxicity: implications for preventive treatments of ischemic stroke and late-onset Alzheimer's disease
Abstract
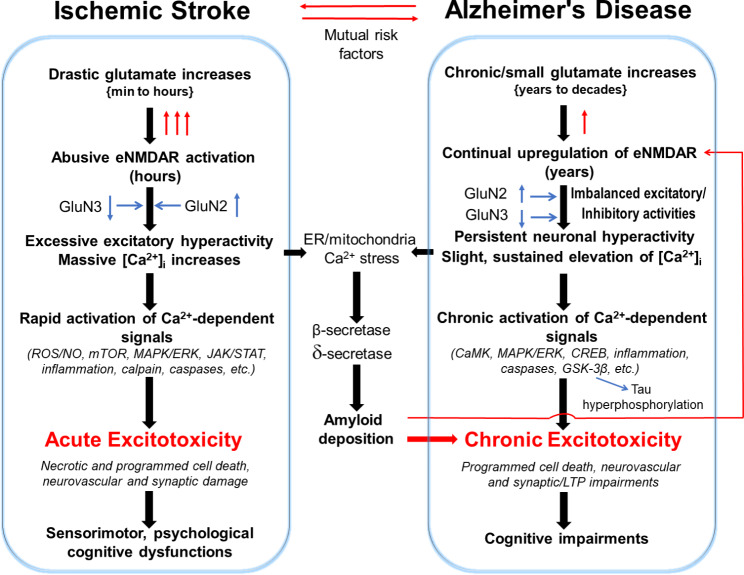
Stroke and late-onset Alzheimer's disease (AD) are risk factors for each other; the comorbidity of these brain disorders in aging individuals represents a significant challenge in basic research and clinical practice. The similarities and differences between stroke and AD in terms of pathogenesis and pathophysiology, however, have rarely been comparably reviewed. Here, we discuss the research background and recent progresses that are important and informative for the comorbidity of stroke and late-onset AD and related dementia (ADRD). Glutamatergic NMDA receptor (NMDAR) activity and NMDAR-mediated Ca2+ influx are essential for neuronal function and cell survival. An ischemic insult, however, can cause rapid increases in glutamate concentration and excessive activation of NMDARs, leading to swift Ca2+ overload in neuronal cells and acute excitotoxicity within hours and days. On the other hand, mild upregulation of NMDAR activity, commonly seen in AD animal models and patients, is not immediately cytotoxic. Sustained NMDAR hyperactivity and Ca2+ dysregulation lasting from months to years, nevertheless, can be pathogenic for slowly evolving events, i.e. degenerative excitotoxicity, in the development of AD/ADRD. Specifically, Ca2+ influx mediated by extrasynaptic NMDARs (eNMDARs) and a downstream pathway mediated by transient receptor potential cation channel subfamily M member (TRPM) are primarily responsible for excitotoxicity. On the other hand, the NMDAR subunit GluN3A plays a "gatekeeper" role in NMDAR activity and a neuroprotective role against both acute and chronic excitotoxicity. Thus, ischemic stroke and AD share an NMDAR- and Ca2+-mediated pathogenic mechanism that provides a common receptor target for preventive and possibly disease-modifying therapies. Memantine (MEM) preferentially blocks eNMDARs and was approved by the Federal Drug Administration (FDA) for symptomatic treatment of moderate-to-severe AD with variable efficacy. According to the pathogenic role of eNMDARs, it is conceivable that MEM and other eNMDAR antagonists should be administered much earlier, preferably during the presymptomatic phases of AD/ADRD. This anti-AD treatment could simultaneously serve as a preconditioning strategy against stroke that attacks ≥ 50% of AD patients. Future research on the regulation of NMDARs, enduring control of eNMDARs, Ca2+ homeostasis, and downstream events will provide a promising opportunity to understand and treat the comorbidity of AD/ADRD and stroke.
Keywords: Alzheimer’s disease; Ca2+ homeostasis; Excitotoxicity; Extrasynaptic NMDARs; GluN2B subunit; GluN3A subunit; Glutamate; Ischemic stroke; Memantine; NMDA receptors.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures





References
-
- Imran TF, Posner D, Honerlaw J, Vassy JL, Song RJ, Ho YL, Kittner SJ, Liao KP, Cai T, O’Donnell CJ, et al. A phenotyping algorithm to identify acute ischemic stroke accurately from a national biobank: the million veteran program. Clin Epidemiol. 2018;10:1509–21. doi: 10.2147/CLEP.S160764. - DOI - PMC - PubMed
-
- Association A. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement 2020, PMID: 32157811. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
