

Analysis of neutral mutational drift in an allosteric enzyme
- PMID: 37402140
- PMCID: PMC10364584
- DOI: 10.1002/pro.4719
Analysis of neutral mutational drift in an allosteric enzyme
Abstract
Neutral mutational drift is an important source of biological diversity that remains underexploited in fundamental studies of protein biophysics. This study uses a synthetic transcriptional circuit to study neutral drift in protein tyrosine phosphatase 1B (PTP1B), a mammalian signaling enzyme for which conformational changes are rate limiting. Kinetic assays of purified mutants indicate that catalytic activity, rather than thermodynamic stability, guides enrichment under neutral drift, where neutral or mildly activating mutations can mitigate the effects of deleterious ones. In general, mutants show a moderate activity-stability tradeoff, an indication that minor improvements in the activity of PTP1B do not require concomitant losses in its stability. Multiplexed sequencing of large mutant pools suggests that substitutions at allosterically influential sites are purged under biological selection, which enriches for mutations located outside of the active site. Findings indicate that the positional dependence of neutral mutations within drifting populations can reveal the presence of allosteric networks and illustrate an approach for using synthetic transcriptional systems to explore these mutations in regulatory enzymes.
Keywords: neutral drift; protein allostery; protein evolutionary patterns; protein tyrosine phosphatases.
© 2023 The Protein Society.
Conflict of interest statement
Jerome M. Fox is a founder of Think Bioscience, Inc., which develops small‐molecule therapeutics and employs Jerome M. Fox and Levi Kramer, who hold equity interests in the company. Think Bioscience is exploring many drug targets, including protein tyrosine phosphatases such as PTP1B. Michael R. Shirts is an Open Science Fellow at and consultant for Psivant Sciences, and a consultant for Relay Therapeutics.
Figures
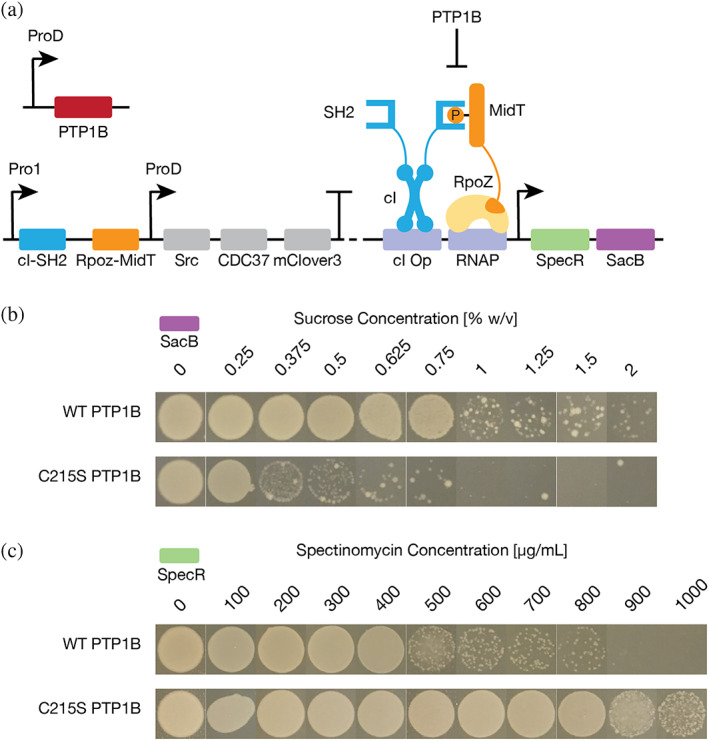
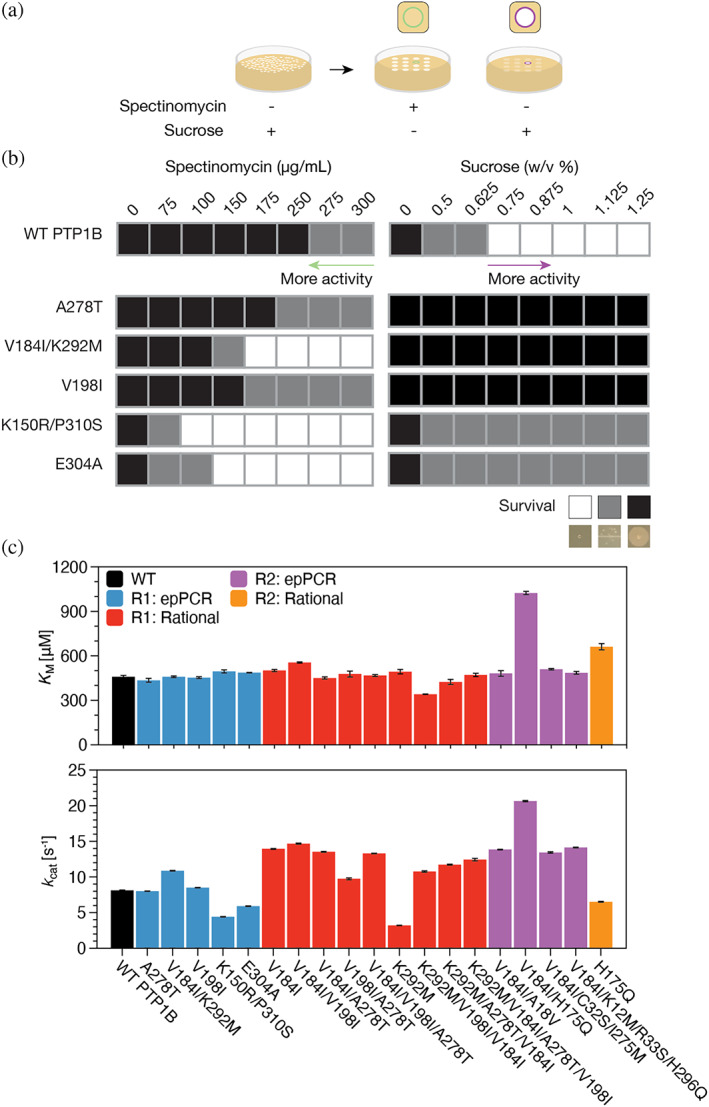
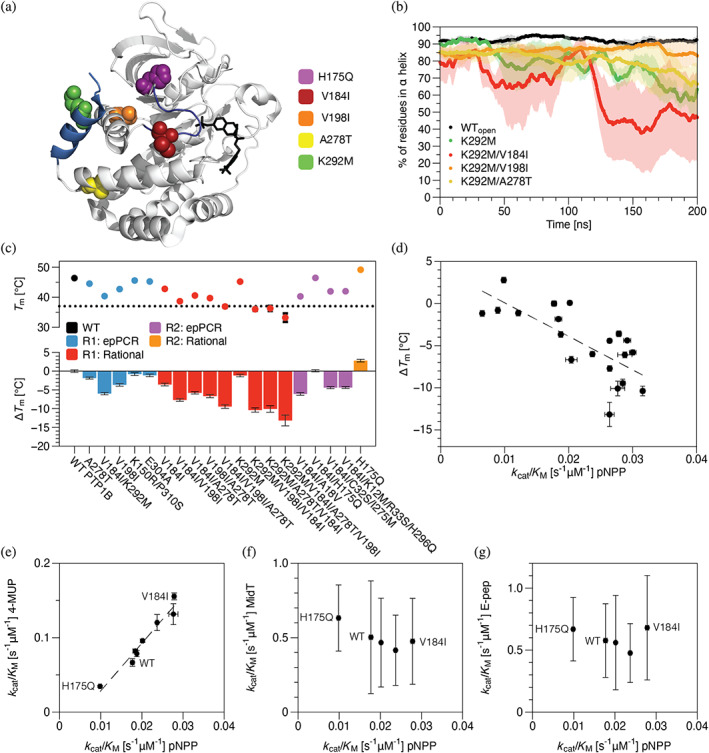
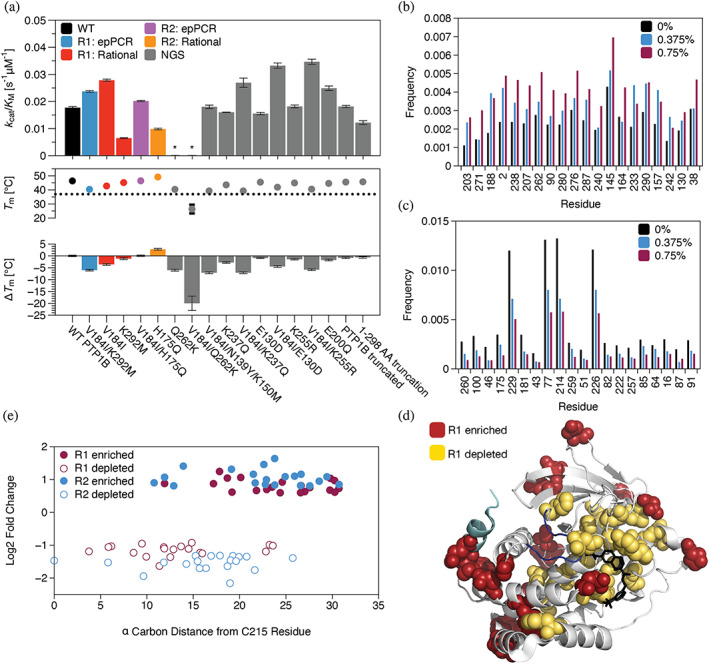
References
-
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. Gromacs: high performance molecular simulations through multi‐level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25.
-
- Arcus VL, Prentice EJ, Hobbs JK, Mulholland AJ, Van Der Kamp MW, Pudney CR, et al. On the temperature dependence of enzyme‐catalyzed rates. Biochemistry. 2016;55:1681–1688. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
