

Targeted high-throughput mutagenesis of the human spliceosome reveals its in vivo operating principles
- PMID: 37402368
- PMCID: PMC10484158
- DOI: 10.1016/j.molcel.2023.06.003
Targeted high-throughput mutagenesis of the human spliceosome reveals its in vivo operating principles
Abstract
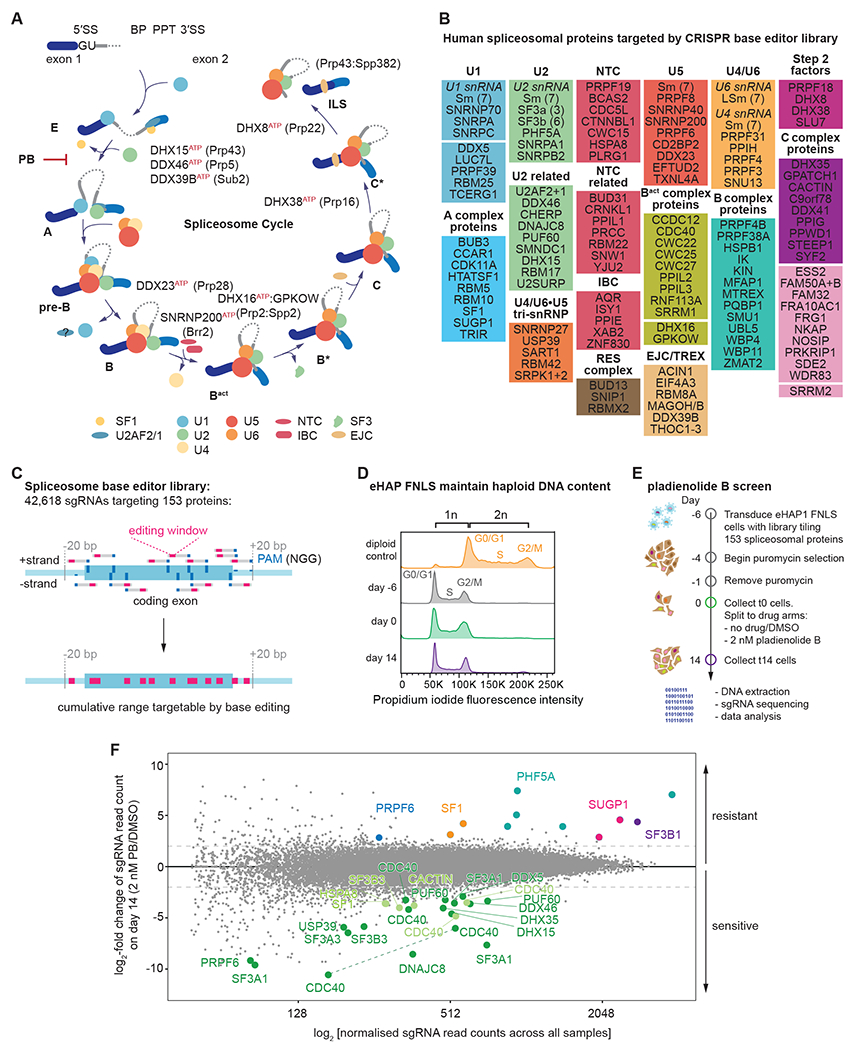
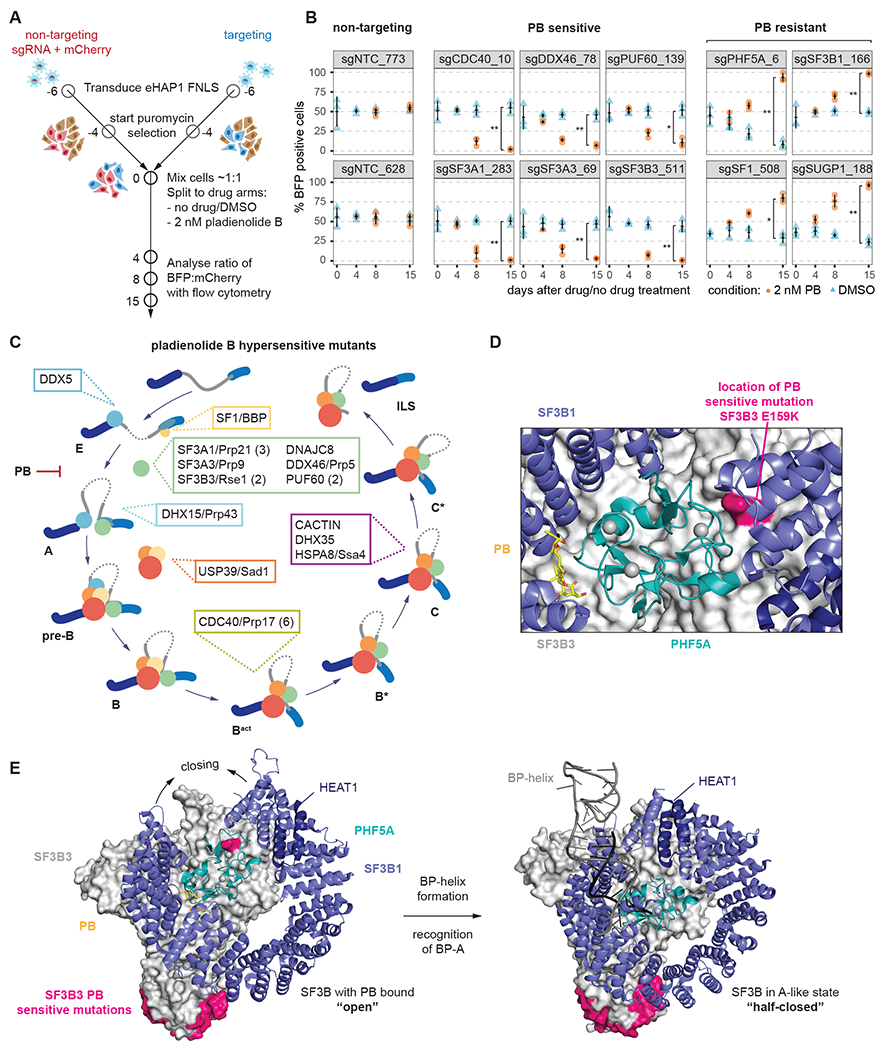
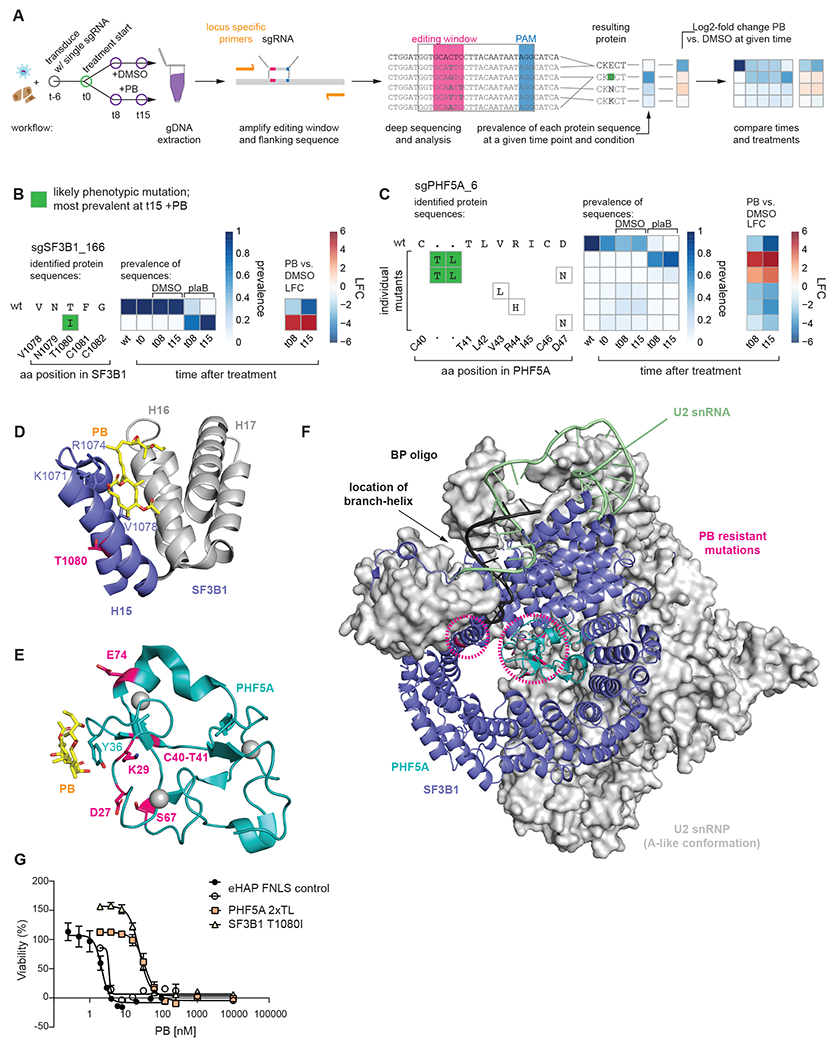
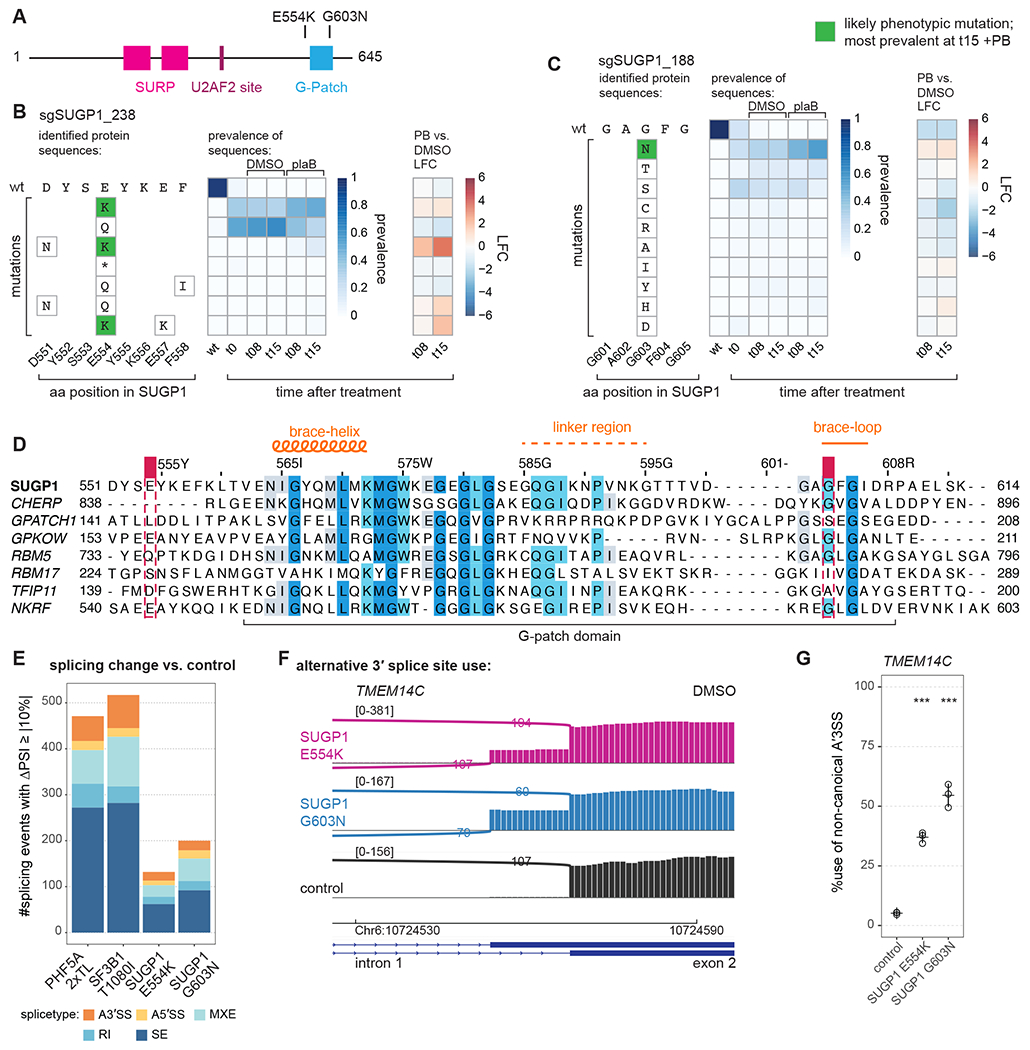
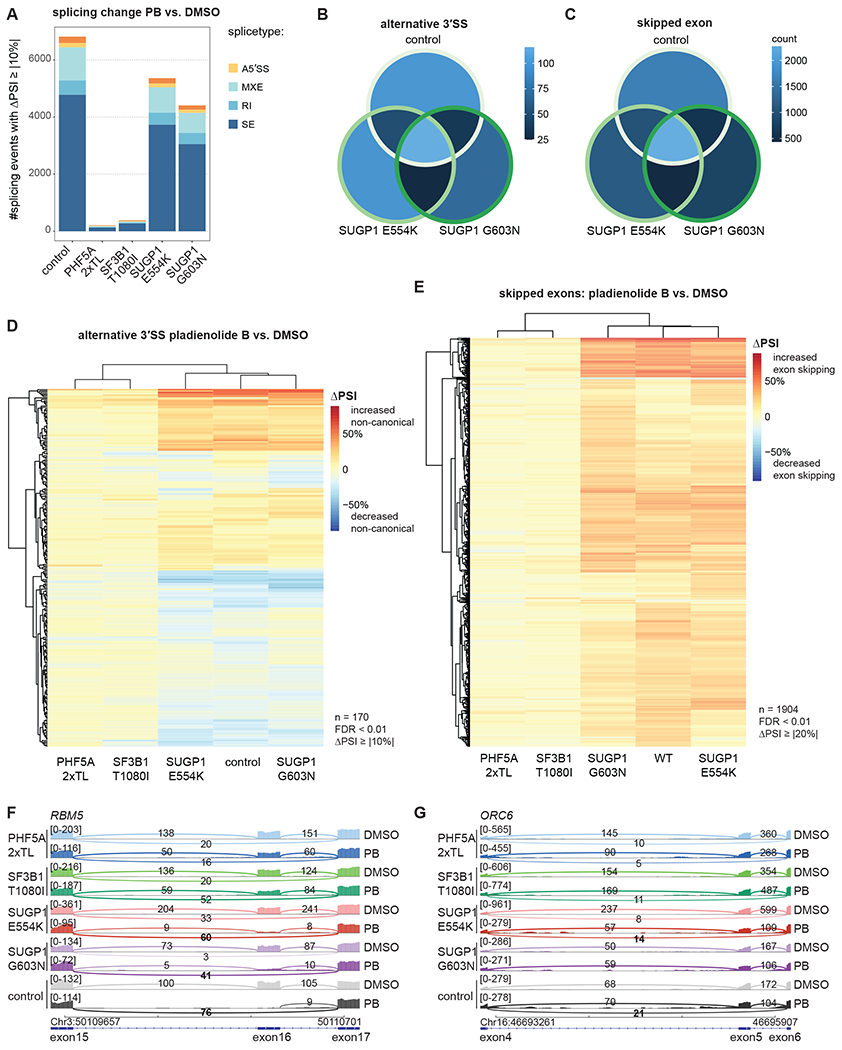
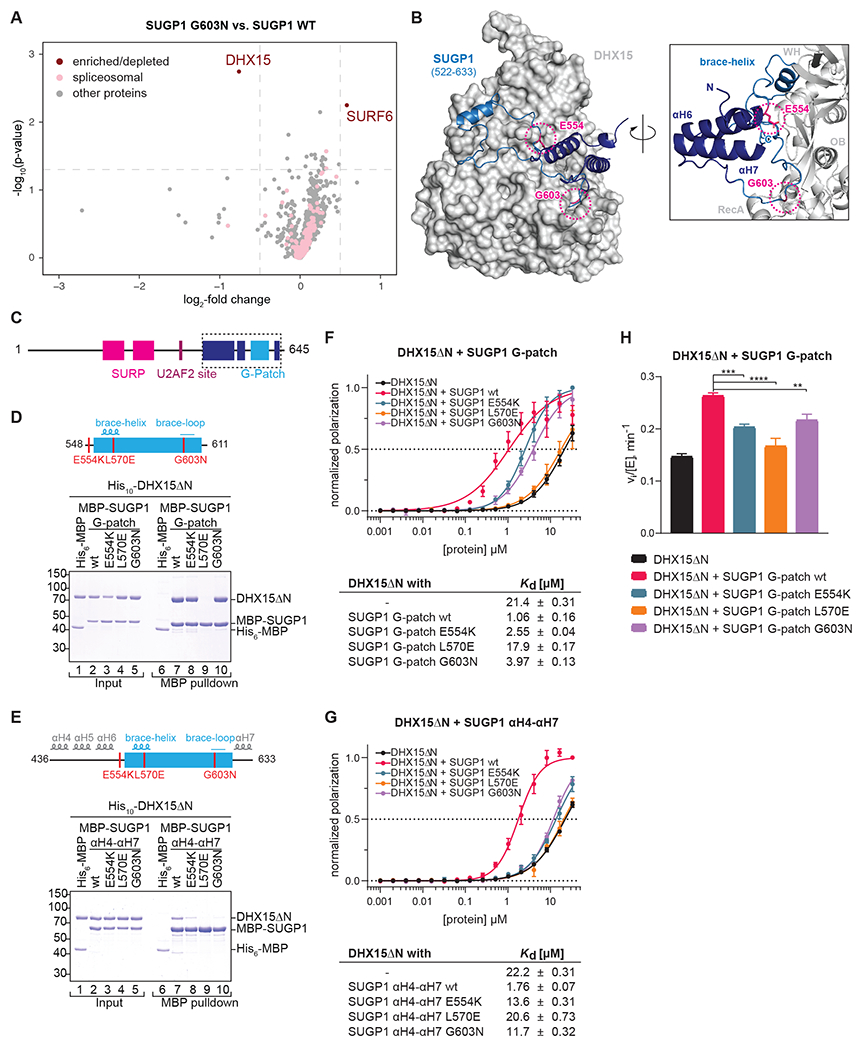
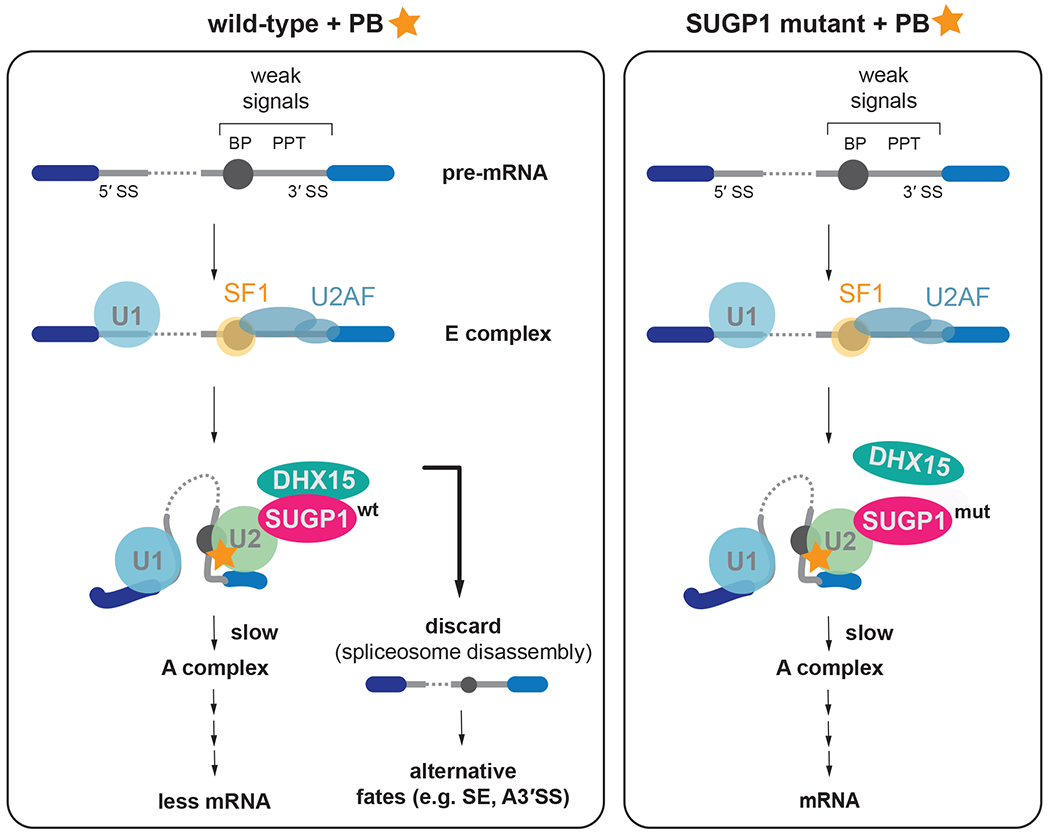
The spliceosome is a staggeringly complex machine, comprising, in humans, 5 snRNAs and >150 proteins. We scaled haploid CRISPR-Cas9 base editing to target the entire human spliceosome and investigated the mutants using the U2 snRNP/SF3b inhibitor, pladienolide B. Hypersensitive substitutions define functional sites in the U1/U2-containing A complex but also in components that act as late as the second chemical step after SF3b is dissociated. Viable resistance substitutions map not only to the pladienolide B-binding site but also to the G-patch domain of SUGP1, which lacks orthologs in yeast. We used these mutants and biochemical approaches to identify the spliceosomal disassemblase DHX15/hPrp43 as the ATPase ligand for SUGP1. These and other data support a model in which SUGP1 promotes splicing fidelity by triggering early spliceosome disassembly in response to kinetic blocks. Our approach provides a template for the analysis of essential cellular machines in humans.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Anzalone AV, Koblan LW, and Liu DR (2020). Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38, 824–844. - PubMed
-
- Bejar R (2016). Splicing Factor Mutations in Cancer. Advances in experimental medicine and biology 907, 215–228. - PubMed
-
- Blencowe BJ (2017). The Relationship between Alternative Splicing and Proteomic Complexity. Trends in biochemical sciences 42, 407–408. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
