

ACE2-EGFR-MAPK signaling contributes to SARS-CoV-2 infection
- PMID: 37402592
- PMCID: PMC10320016
- DOI: 10.26508/lsa.202201880
ACE2-EGFR-MAPK signaling contributes to SARS-CoV-2 infection
Abstract
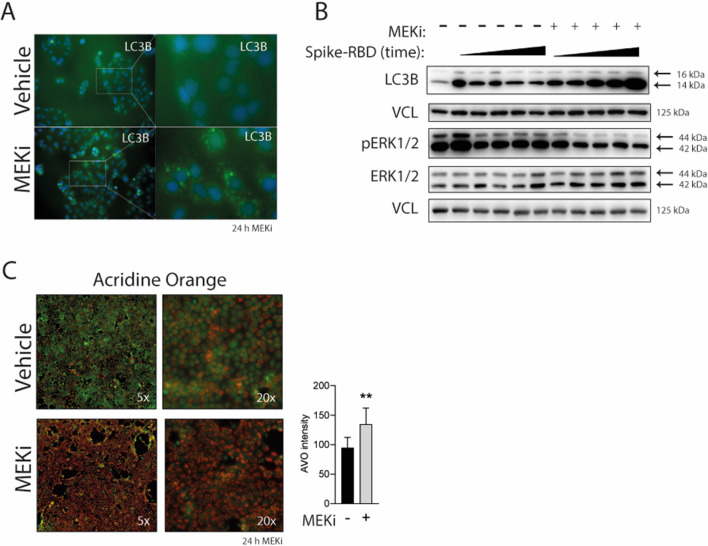
SARS-CoV-2 triggered the most severe pandemic of recent times. To enter into a host cell, SARS-CoV-2 binds to the angiotensin-converting enzyme 2 (ACE2). However, subsequent studies indicated that other cell membrane receptors may act as virus-binding partners. Among these receptors, the epidermal growth factor receptor (EGFR) was hypothesized not only as a spike protein binder, but also to be activated in response to SARS-CoV-2. In our study, we aim at dissecting EGFR activation and its major downstream signaling pathway, the mitogen-activated signaling pathway (MAPK), in SARS-CoV-2 infection. Here, we demonstrate the activation of EGFR-MAPK signaling axis by the SARS-CoV-2 spike protein and we identify a yet unknown cross talk between ACE2 and EGFR that regulated ACE2 abundance and EGFR activation and subcellular localization, respectively. By inhibiting the EGFR-MAPK activation, we observe a reduced infection with either spike-pseudotyped particles or authentic SARS-CoV-2, thus indicating that EGFR serves as a cofactor and the activation of EGFR-MAPK contributes to SARS-CoV-2 infection.
© 2023 Engler et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures










References
-
- Albiges L, Foulon S, Bayle A, Gachot B, Pommeret F, Willekens C, Stoclin A, Merad M, Griscelli F, Lacroix L, et al. (2020) Determinants of the outcomes of patients with cancer infected with SARS-CoV-2: Results from the Gustave Roussy cohort. Nat Cancer 1: 965–975. 10.1038/s43018-020-00120-5 - DOI - PubMed
-
- Bestle D, Heindl MR, Limburg H, Van Lam van T, Pilgram O, Moulton H, Stein DA, Hardes K, Eickmann M, Dolnik O, et al. (2020) TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance 3: e202000786. 10.26508/LSA.202000786 - DOI - PMC - PubMed
-
- Blandin AF, Cruz Da Silva E, Mercier MC, Glushonkov O, Didier P, Dedieu S, Schneider C, Devy J, Etienne-Selloum N, Dontenwill M, et al. (2021) Gefitinib induces EGFR and α5β1 integrin co-endocytosis in glioblastoma cells. Cell Mol Life Sci 78: 2949–2962. 10.1007/s00018-020-03686-6 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous