

EMT activates exocytotic Rabs to coordinate invasion and immunosuppression in lung cancer
- PMID: 37406091
- PMCID: PMC10334751
- DOI: 10.1073/pnas.2220276120
EMT activates exocytotic Rabs to coordinate invasion and immunosuppression in lung cancer
Abstract
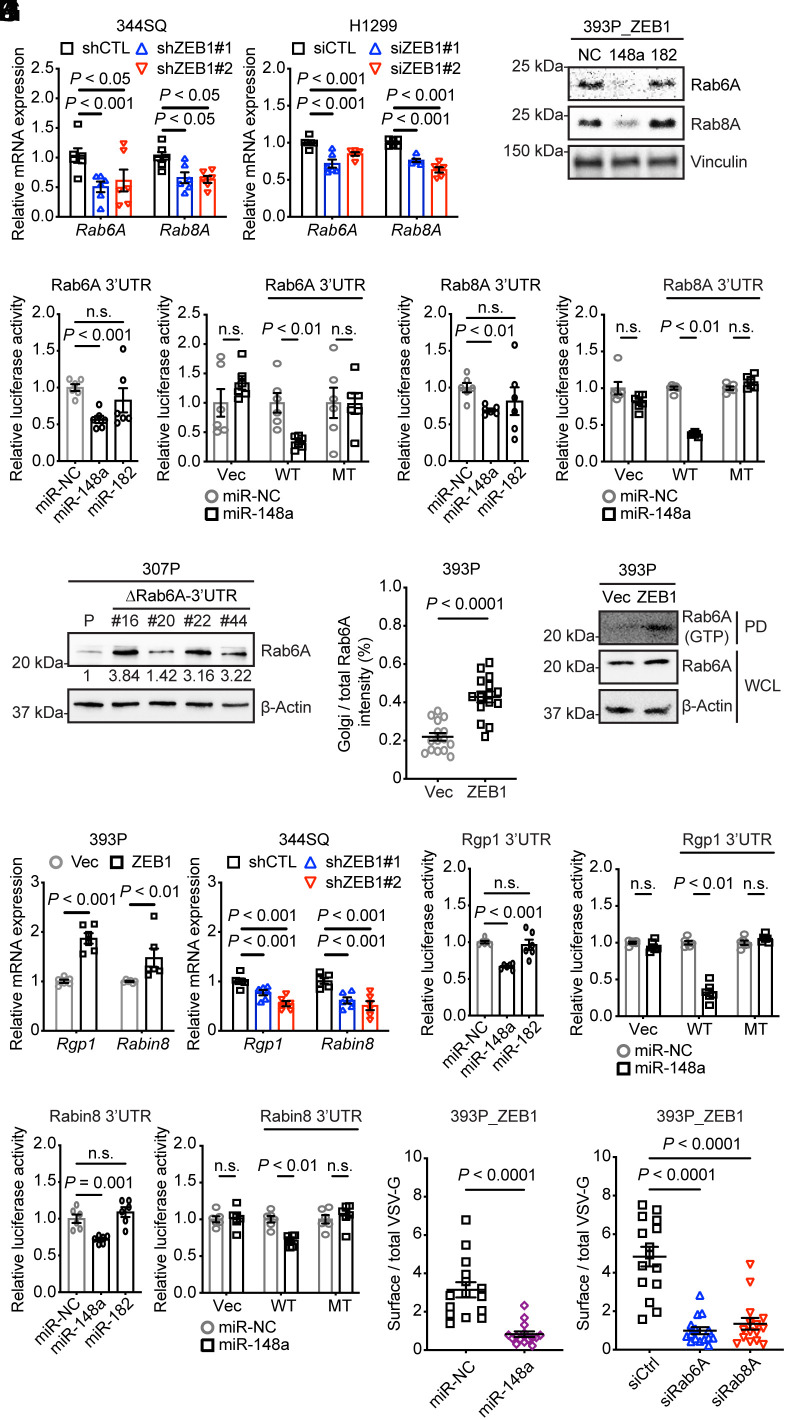
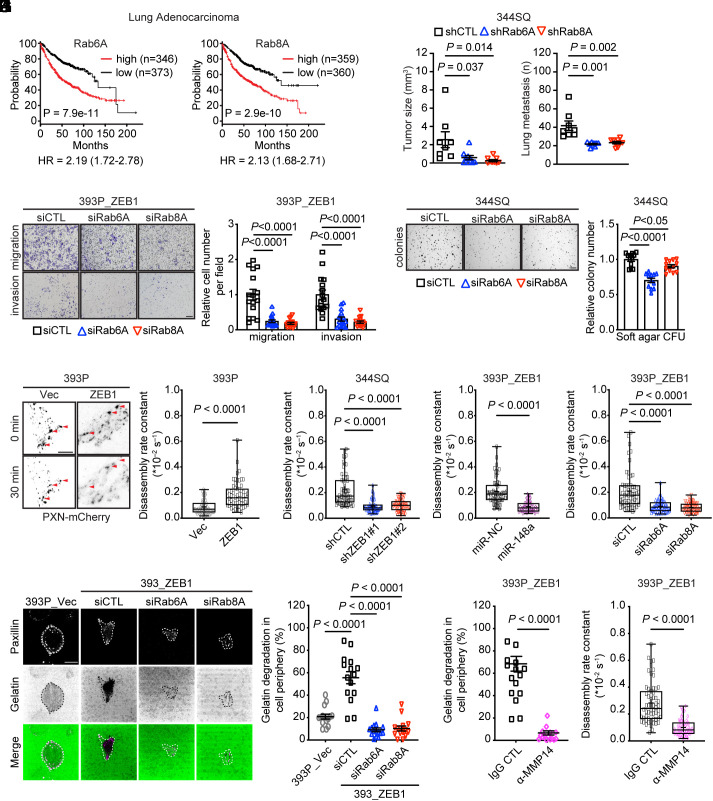
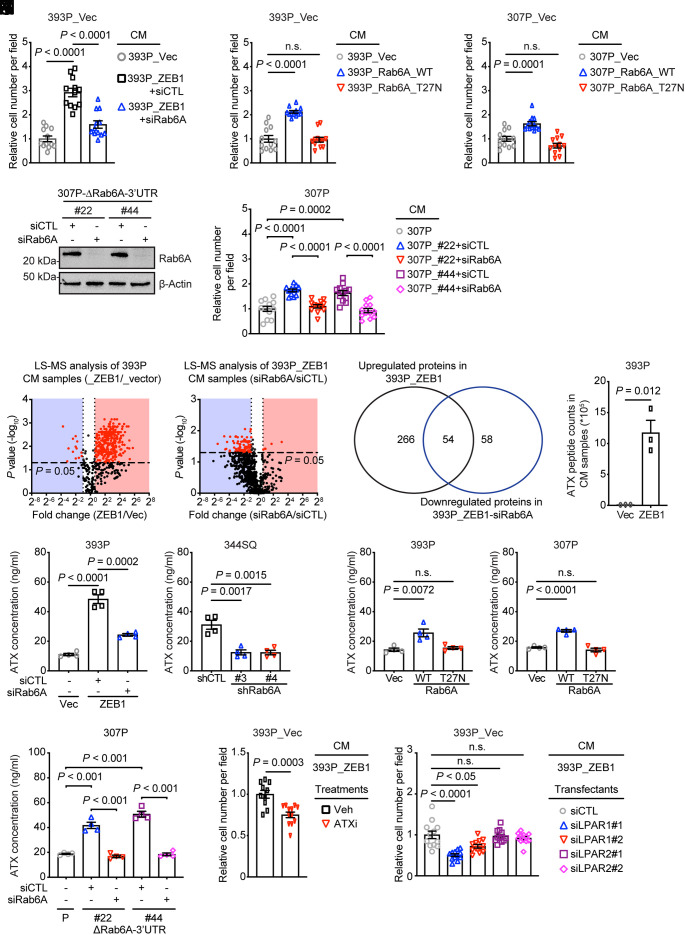
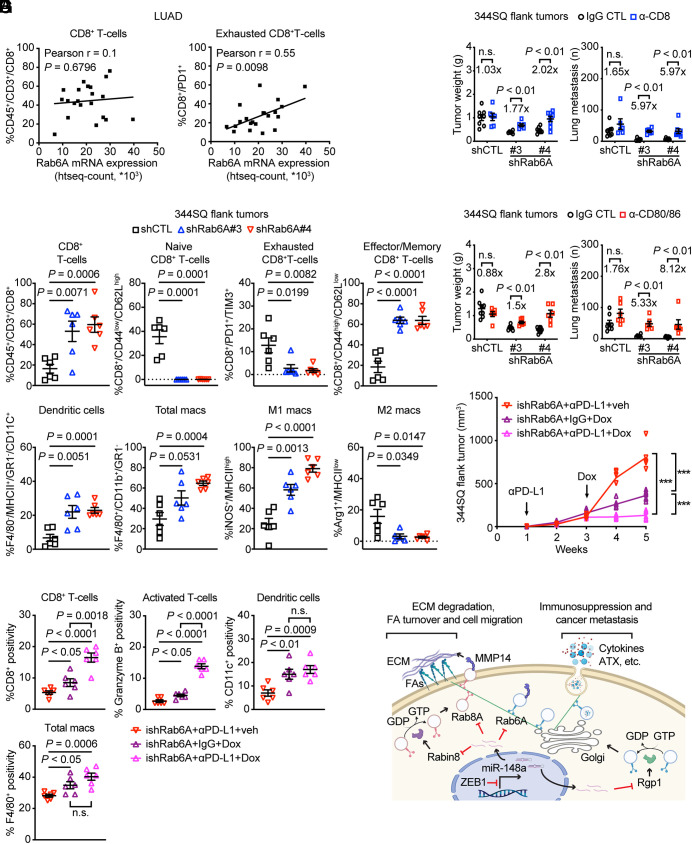
Epithelial-to-mesenchymal transition (EMT) underlies immunosuppression, drug resistance, and metastasis in epithelial malignancies. However, the way in which EMT orchestrates disparate biological processes remains unclear. Here, we identify an EMT-activated vesicular trafficking network that coordinates promigratory focal adhesion dynamics with an immunosuppressive secretory program in lung adenocarcinoma (LUAD). The EMT-activating transcription factor ZEB1 drives exocytotic vesicular trafficking by relieving Rab6A, Rab8A, and guanine nucleotide exchange factors from miR-148a-dependent silencing, thereby facilitating MMP14-dependent focal adhesion turnover in LUAD cells and autotaxin-mediated CD8+ T cell exhaustion, indicating that cell-intrinsic and extrinsic processes are linked through a microRNA that coordinates vesicular trafficking networks. Blockade of ZEB1-dependent secretion reactivates antitumor immunity and negates resistance to PD-L1 immune checkpoint blockade, an important clinical problem in LUAD. Thus, EMT activates exocytotic Rabs to drive a secretory program that promotes invasion and immunosuppression in LUAD.
Keywords: epithelial-mesenchymal transition (EMT); lung cancer; membrane trafficking.
Conflict of interest statement
J.M.K. has received consulting fees from Halozyme.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
