

Cooperation between bHLH transcription factors and histones for DNA access
- PMID: 37407816
- PMCID: PMC10338342
- DOI: 10.1038/s41586-023-06282-3
Cooperation between bHLH transcription factors and histones for DNA access
Abstract
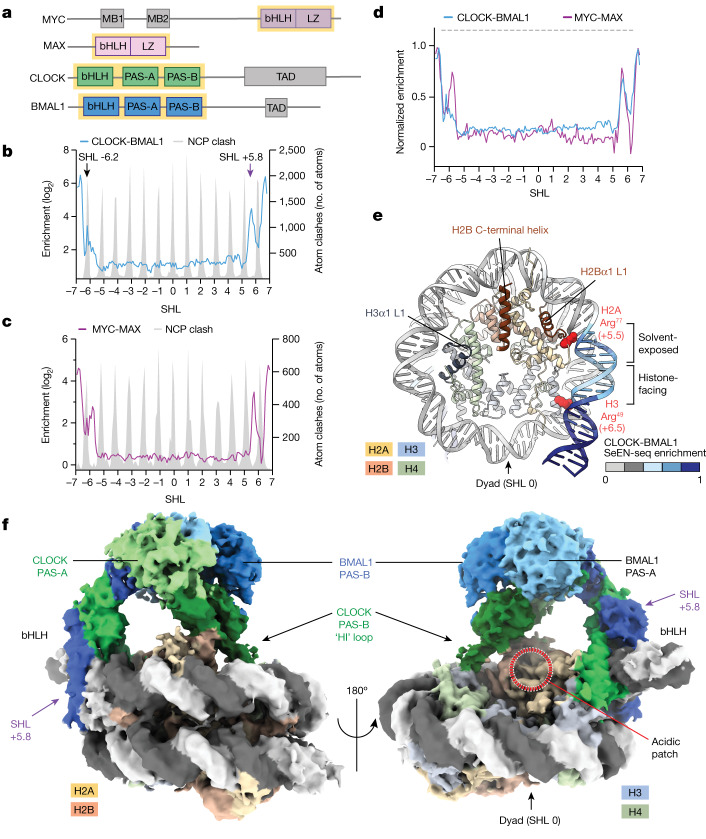
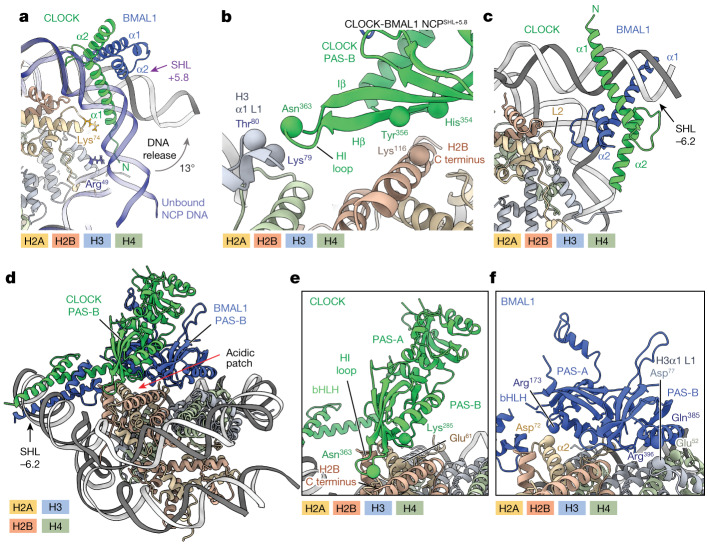
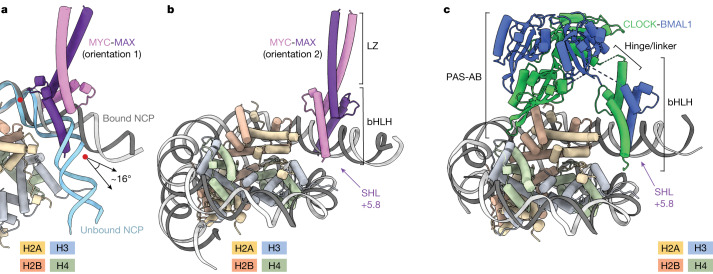
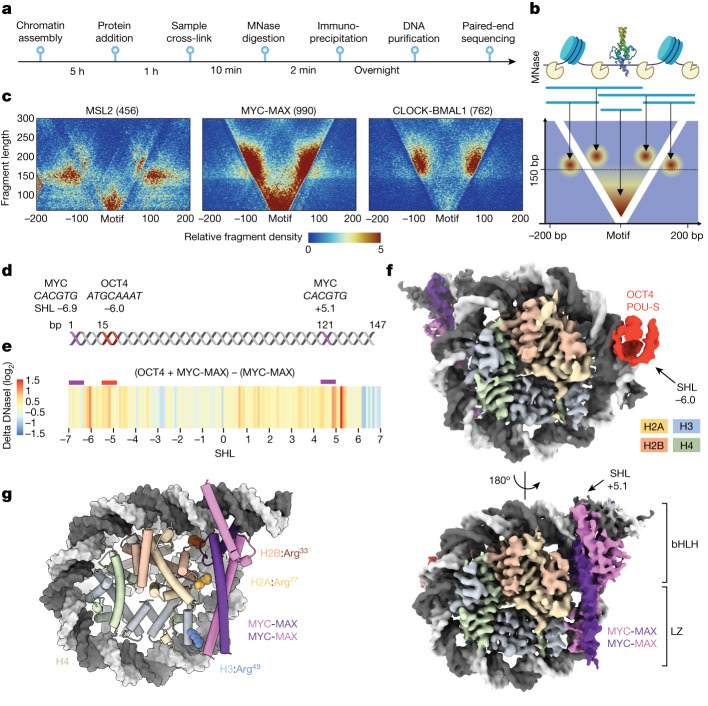
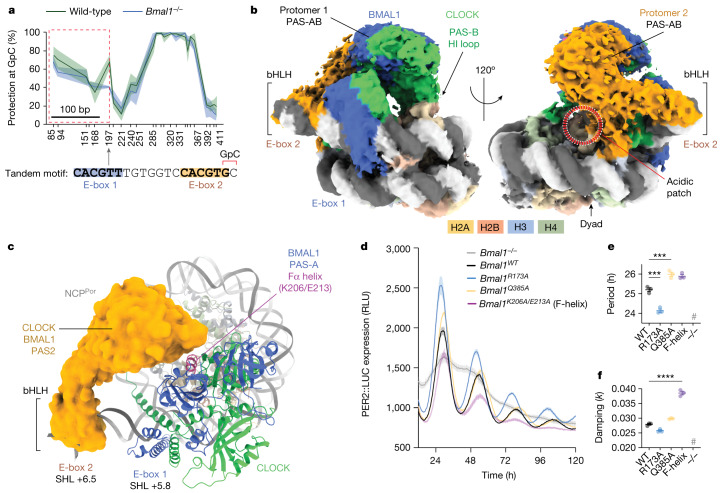
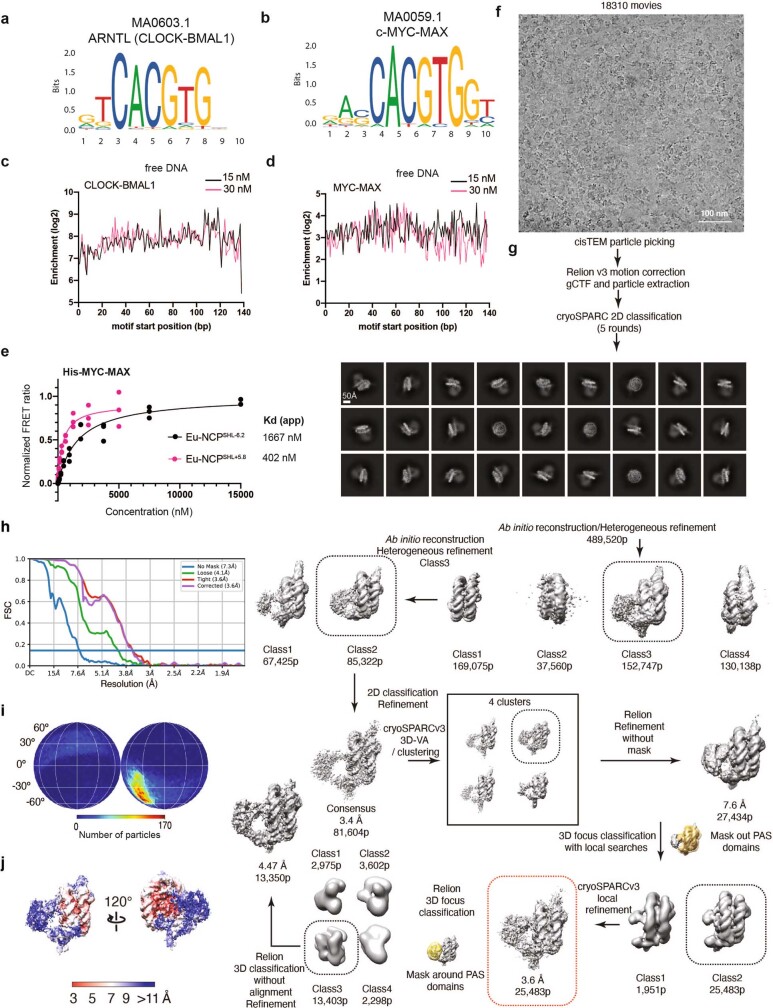
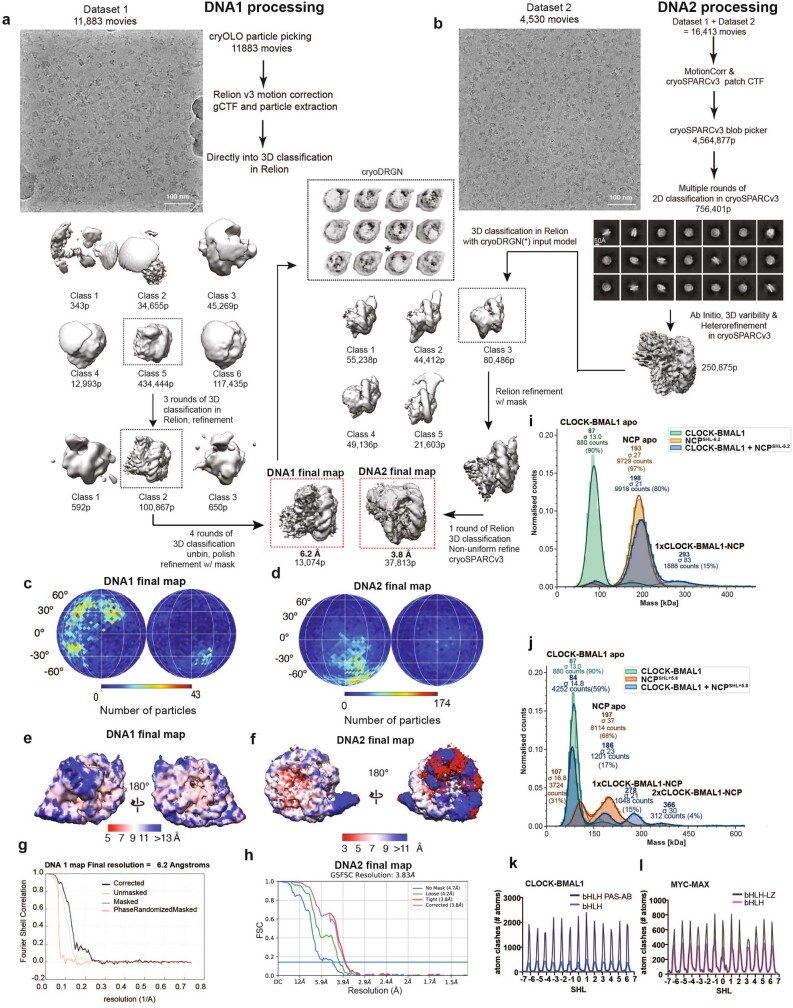
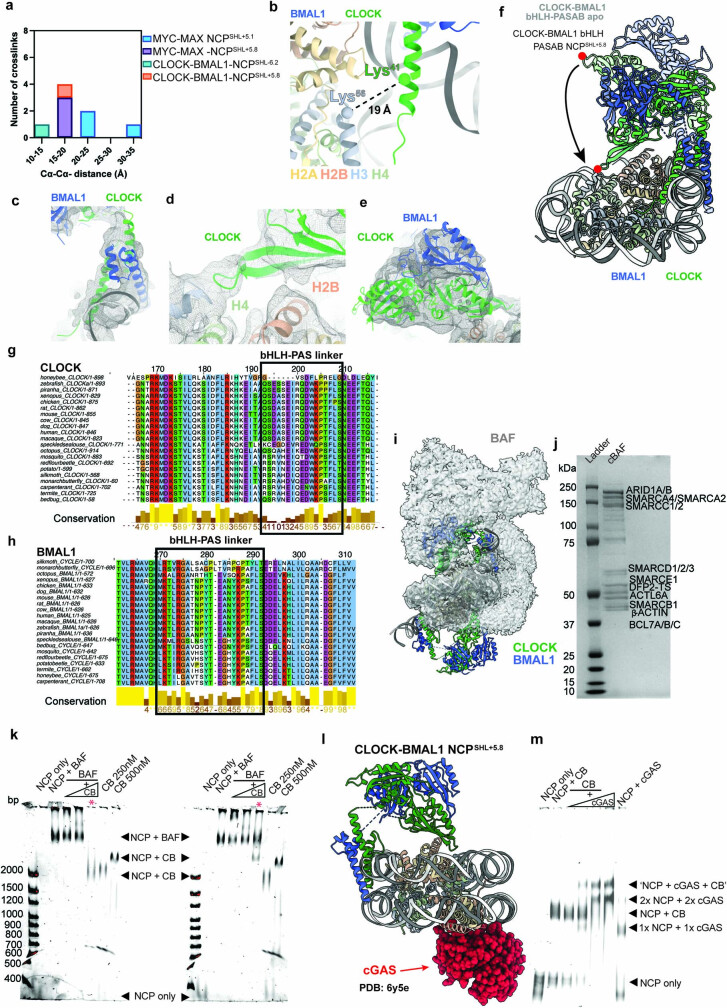
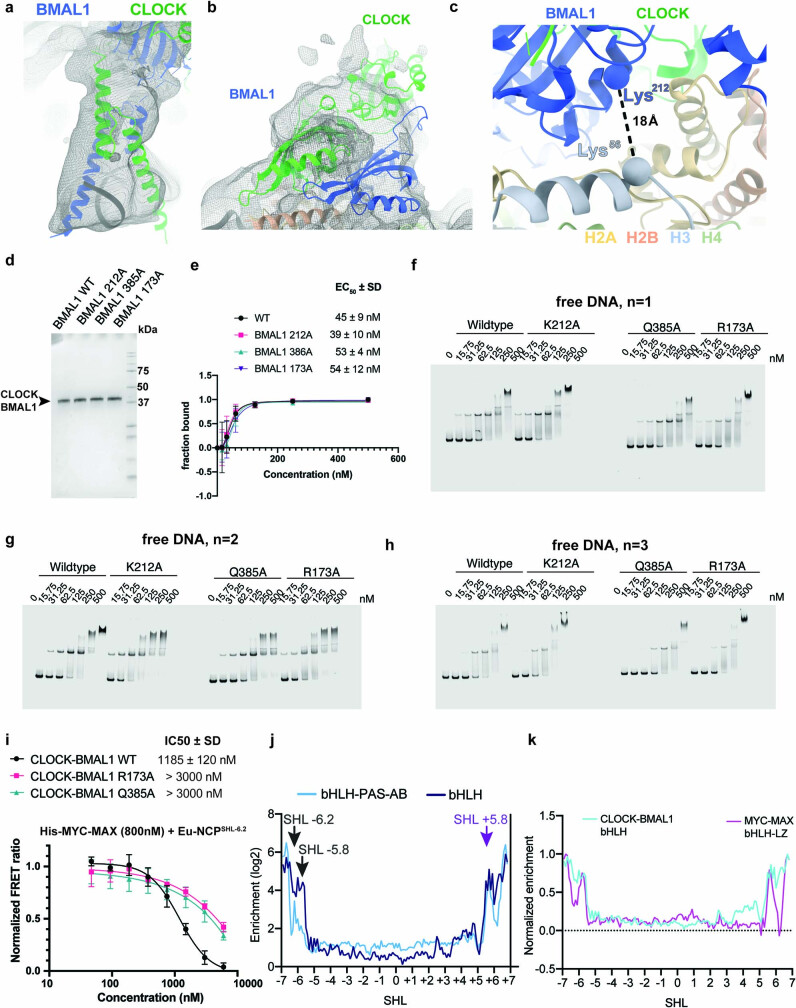
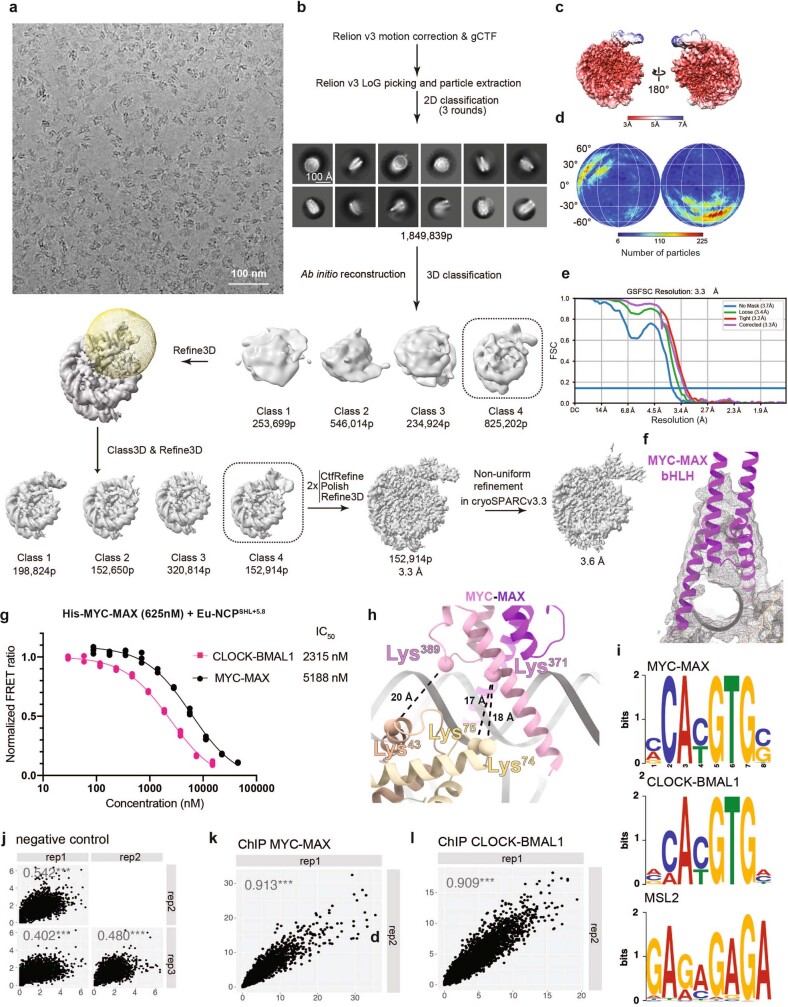
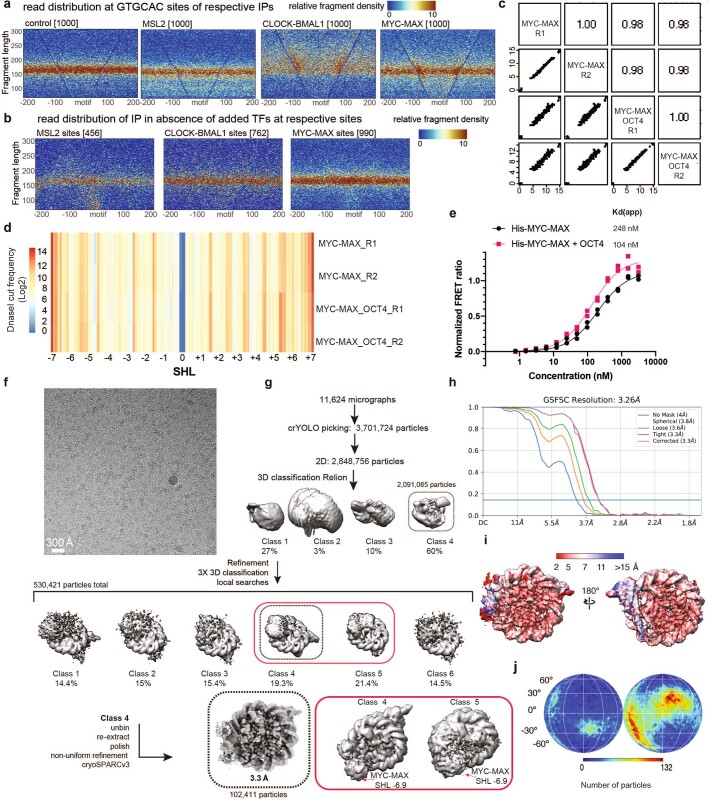
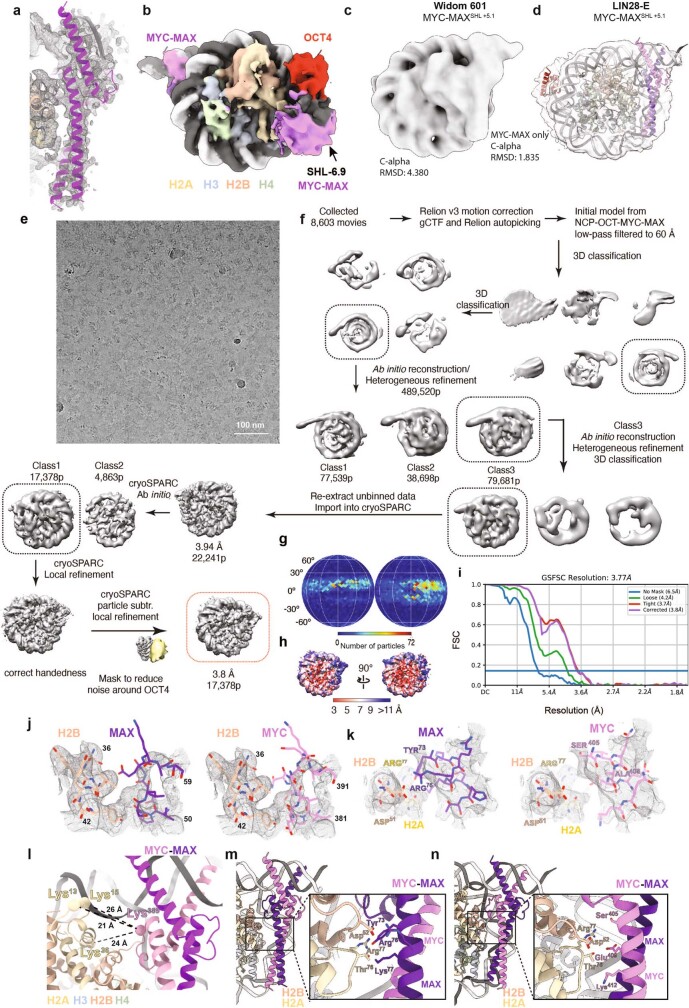
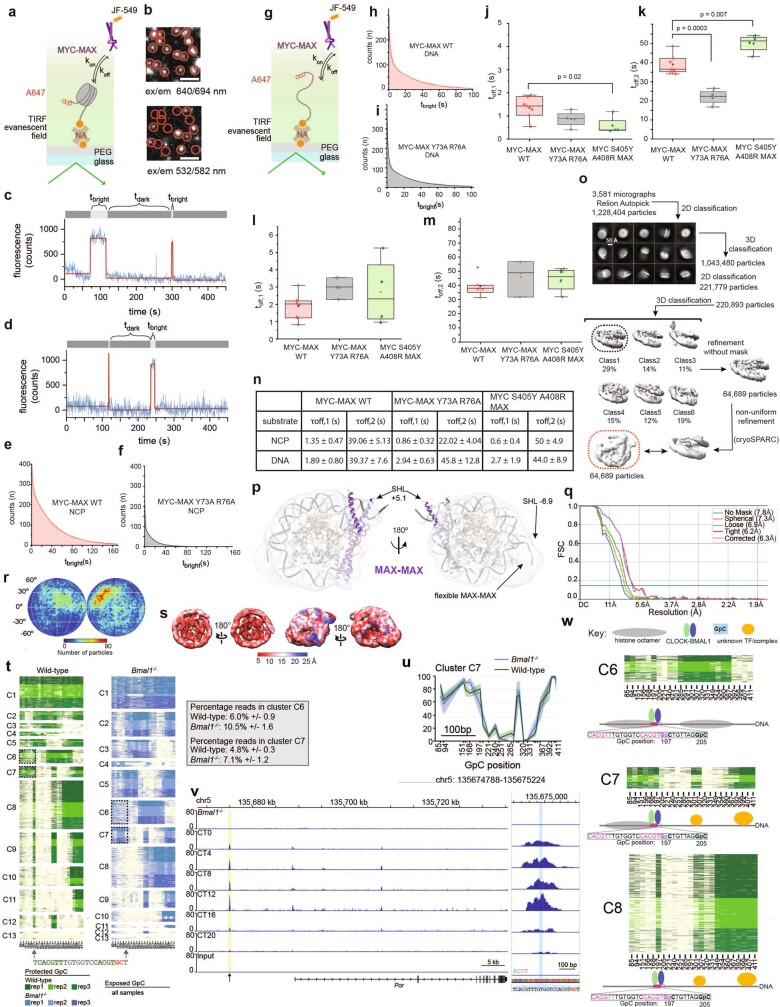
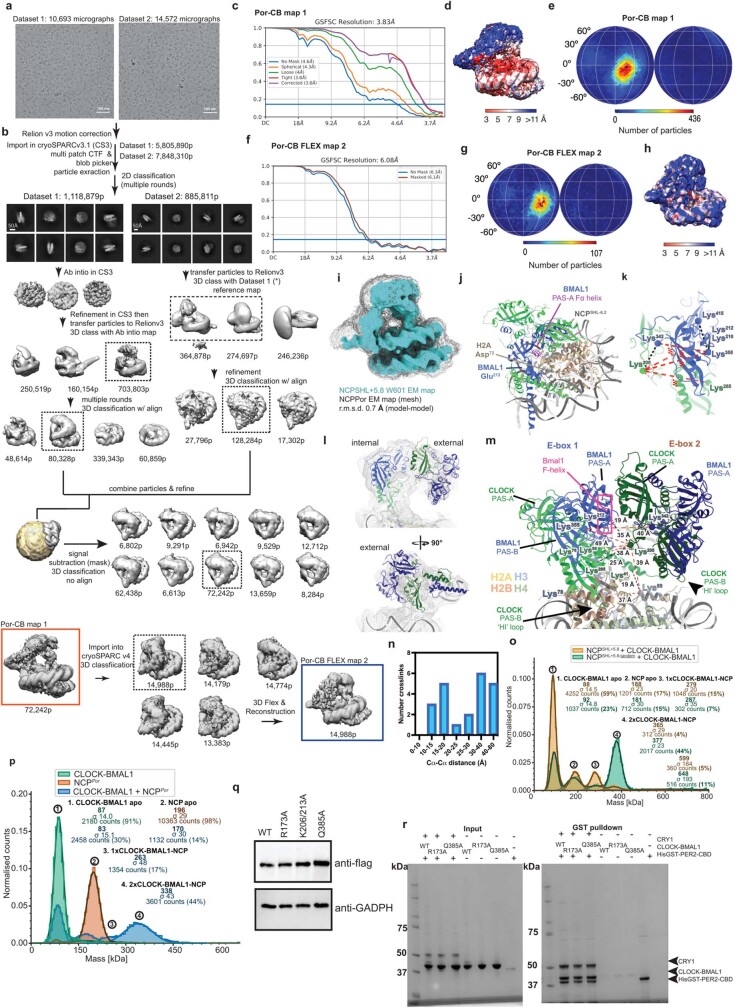
The basic helix-loop-helix (bHLH) family of transcription factors recognizes DNA motifs known as E-boxes (CANNTG) and includes 108 members1. Here we investigate how chromatinized E-boxes are engaged by two structurally diverse bHLH proteins: the proto-oncogene MYC-MAX and the circadian transcription factor CLOCK-BMAL1 (refs. 2,3). Both transcription factors bind to E-boxes preferentially near the nucleosomal entry-exit sites. Structural studies with engineered or native nucleosome sequences show that MYC-MAX or CLOCK-BMAL1 triggers the release of DNA from histones to gain access. Atop the H2A-H2B acidic patch4, the CLOCK-BMAL1 Per-Arnt-Sim (PAS) dimerization domains engage the histone octamer disc. Binding of tandem E-boxes5-7 at endogenous DNA sequences occurs through direct interactions between two CLOCK-BMAL1 protomers and histones and is important for circadian cycling. At internal E-boxes, the MYC-MAX leucine zipper can also interact with histones H2B and H3, and its binding is indirectly enhanced by OCT4 elsewhere on the nucleosome. The nucleosomal E-box position and the type of bHLH dimerization domain jointly determine the histone contact, the affinity and the degree of competition and cooperativity with other nucleosome-bound factors.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
