

Therapy-induced APOBEC3A drives evolution of persistent cancer cells
- PMID: 37407818
- PMCID: PMC10804446
- DOI: 10.1038/s41586-023-06303-1
Therapy-induced APOBEC3A drives evolution of persistent cancer cells
Abstract
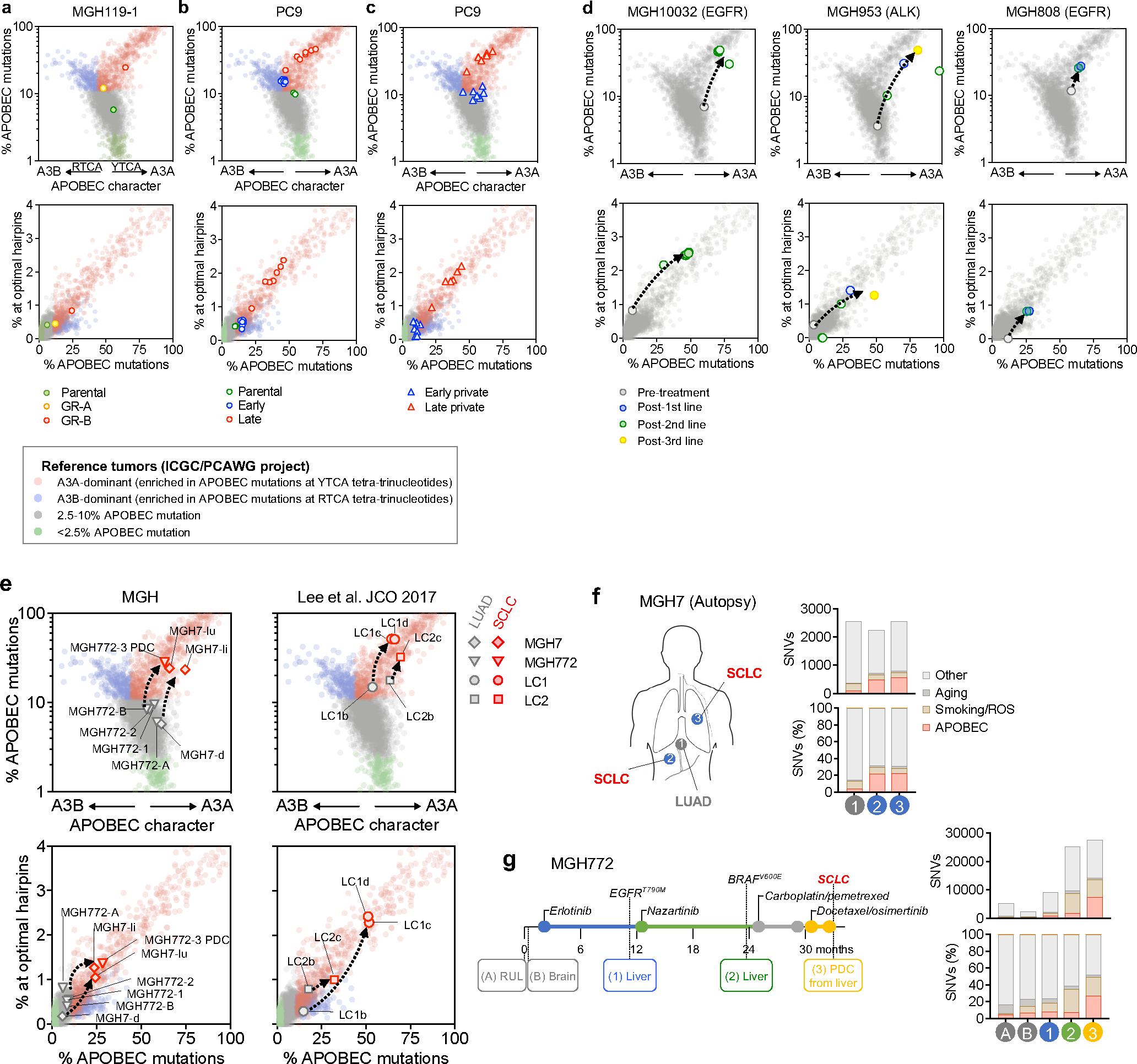
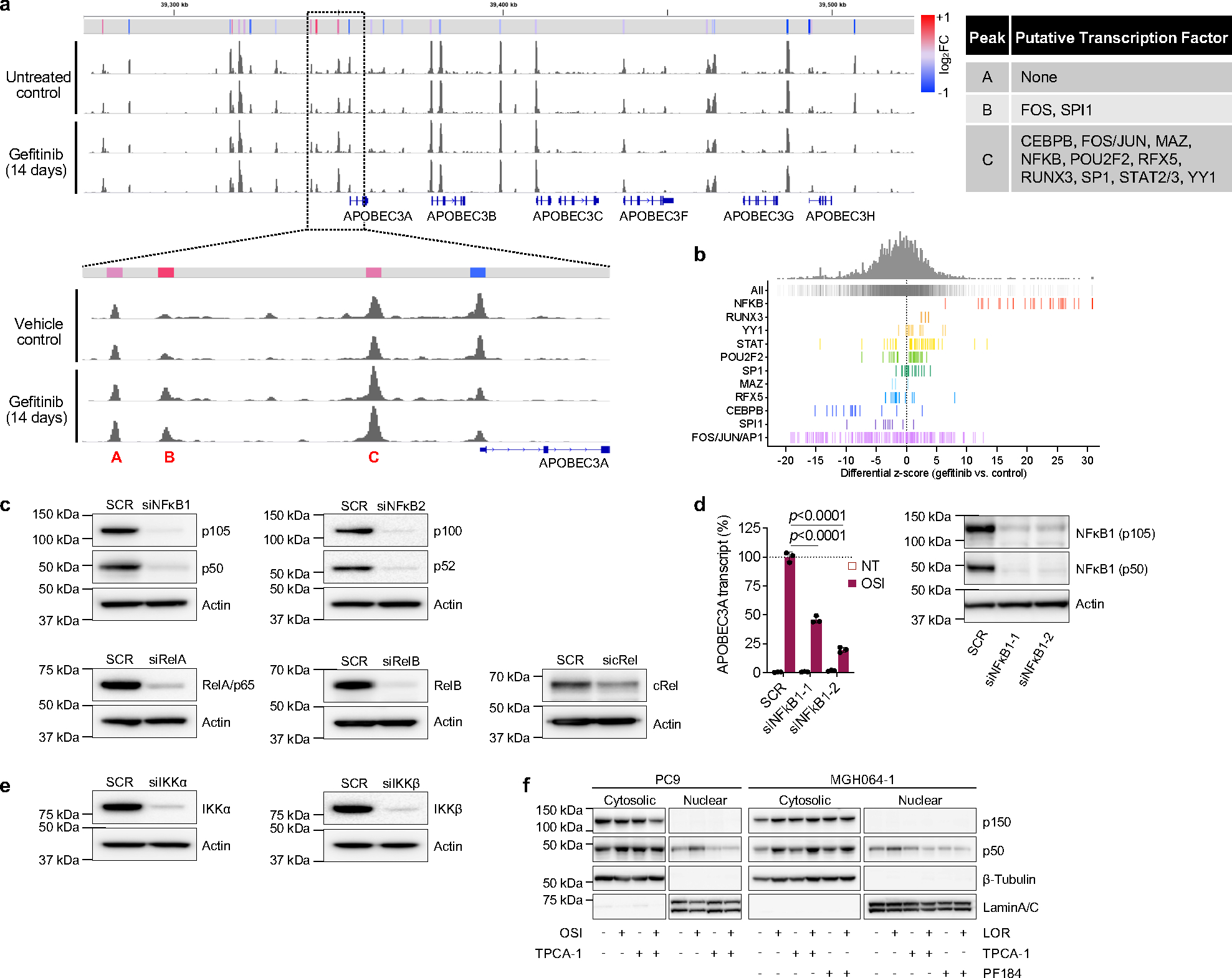
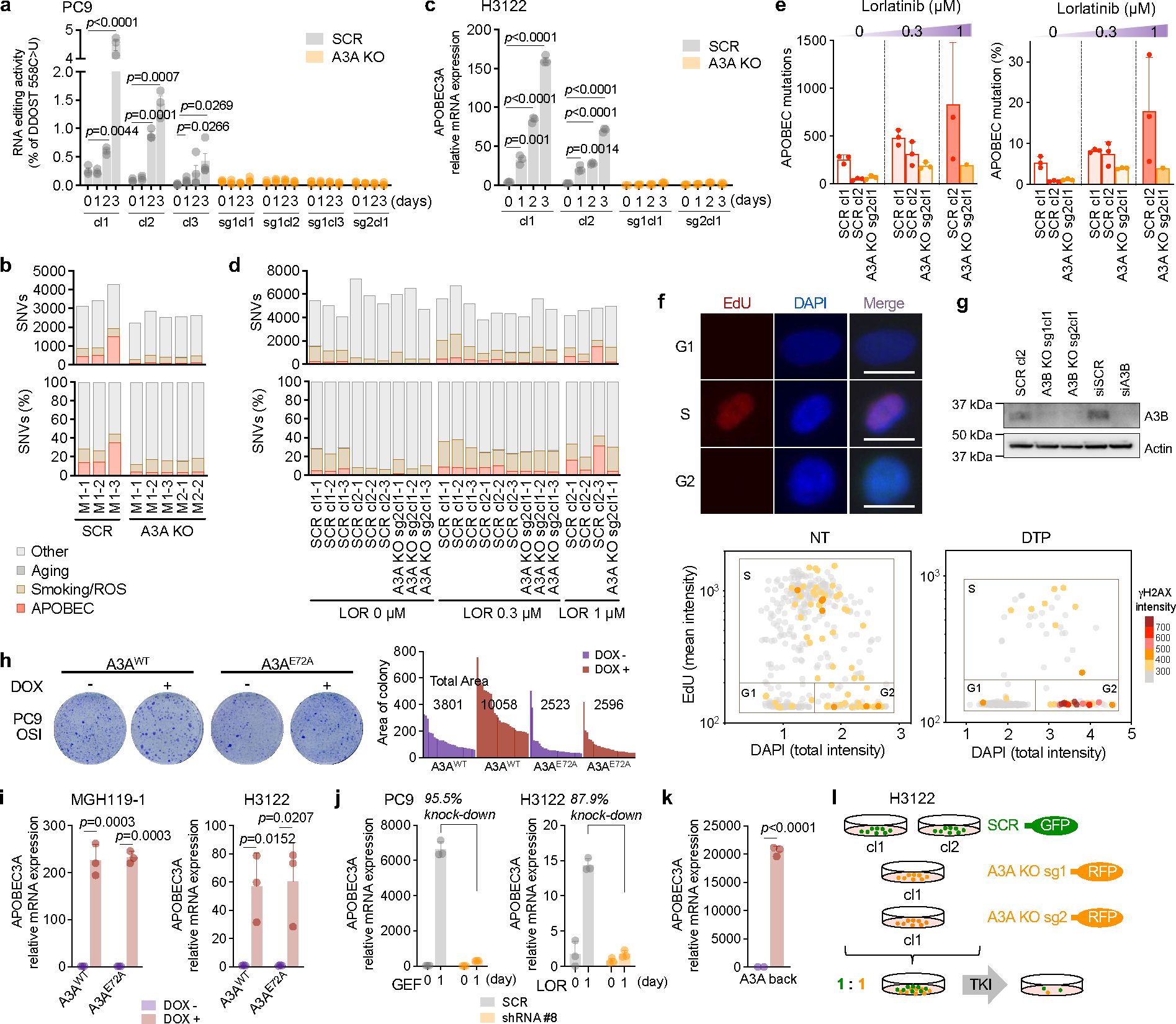
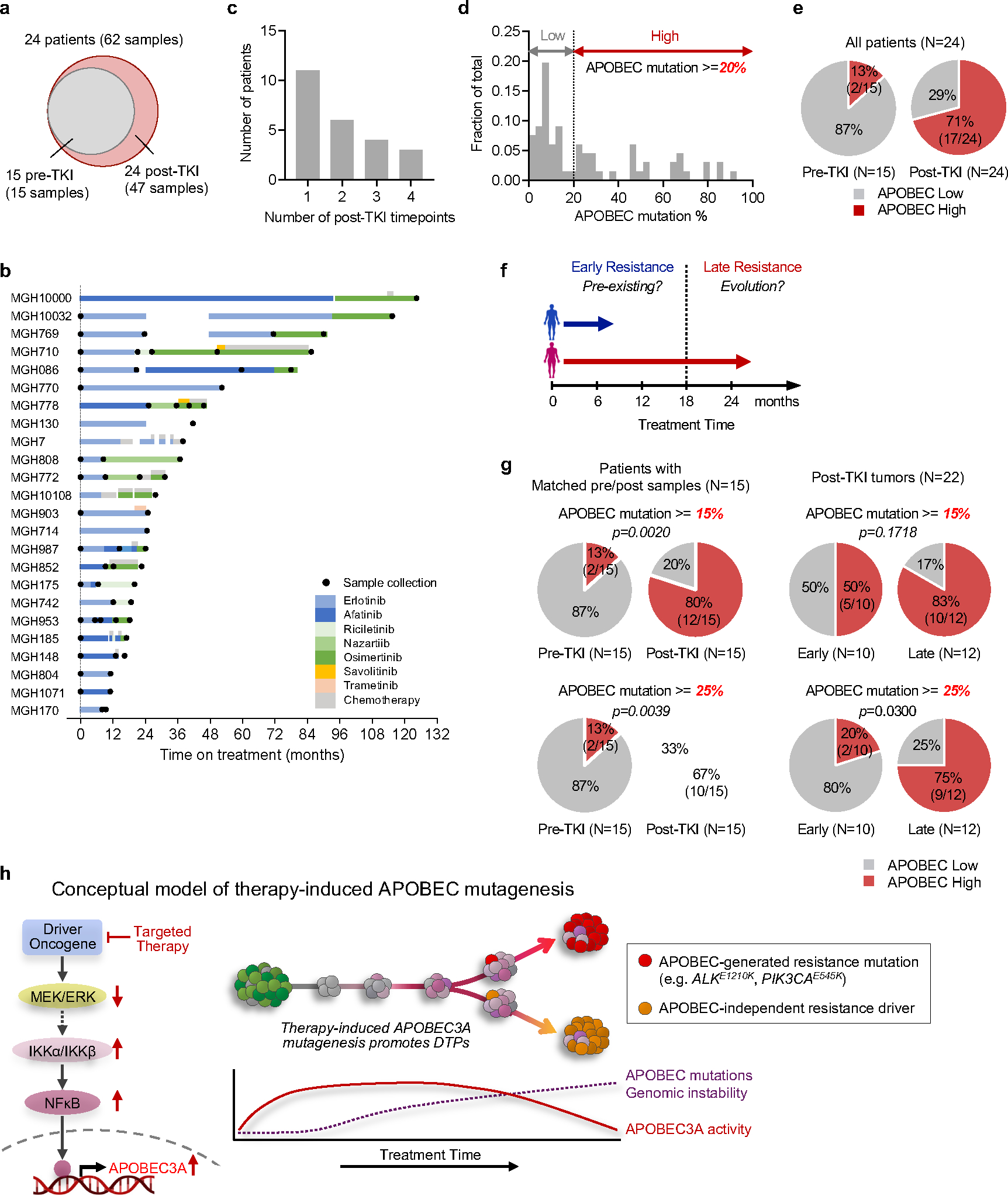
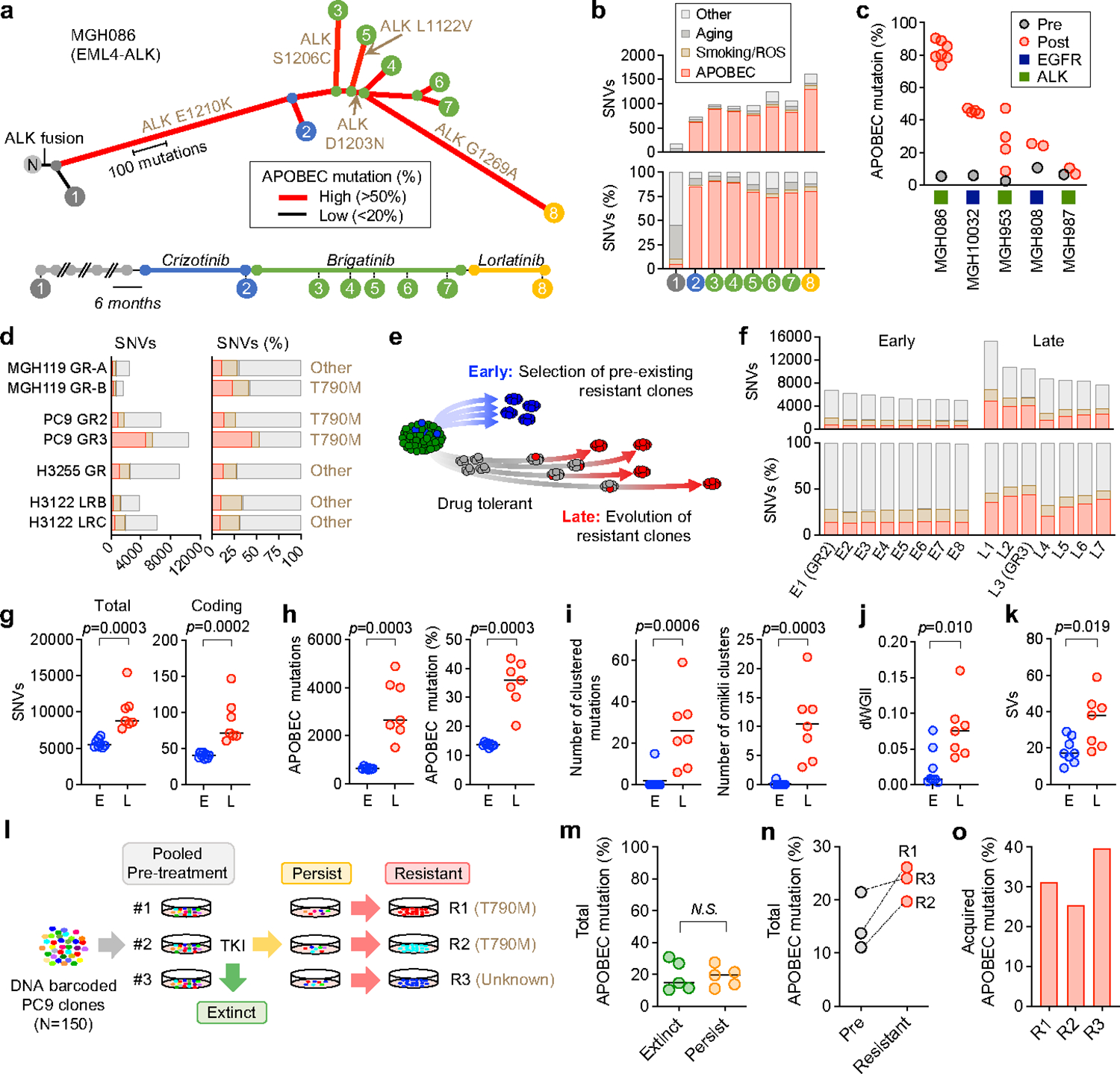
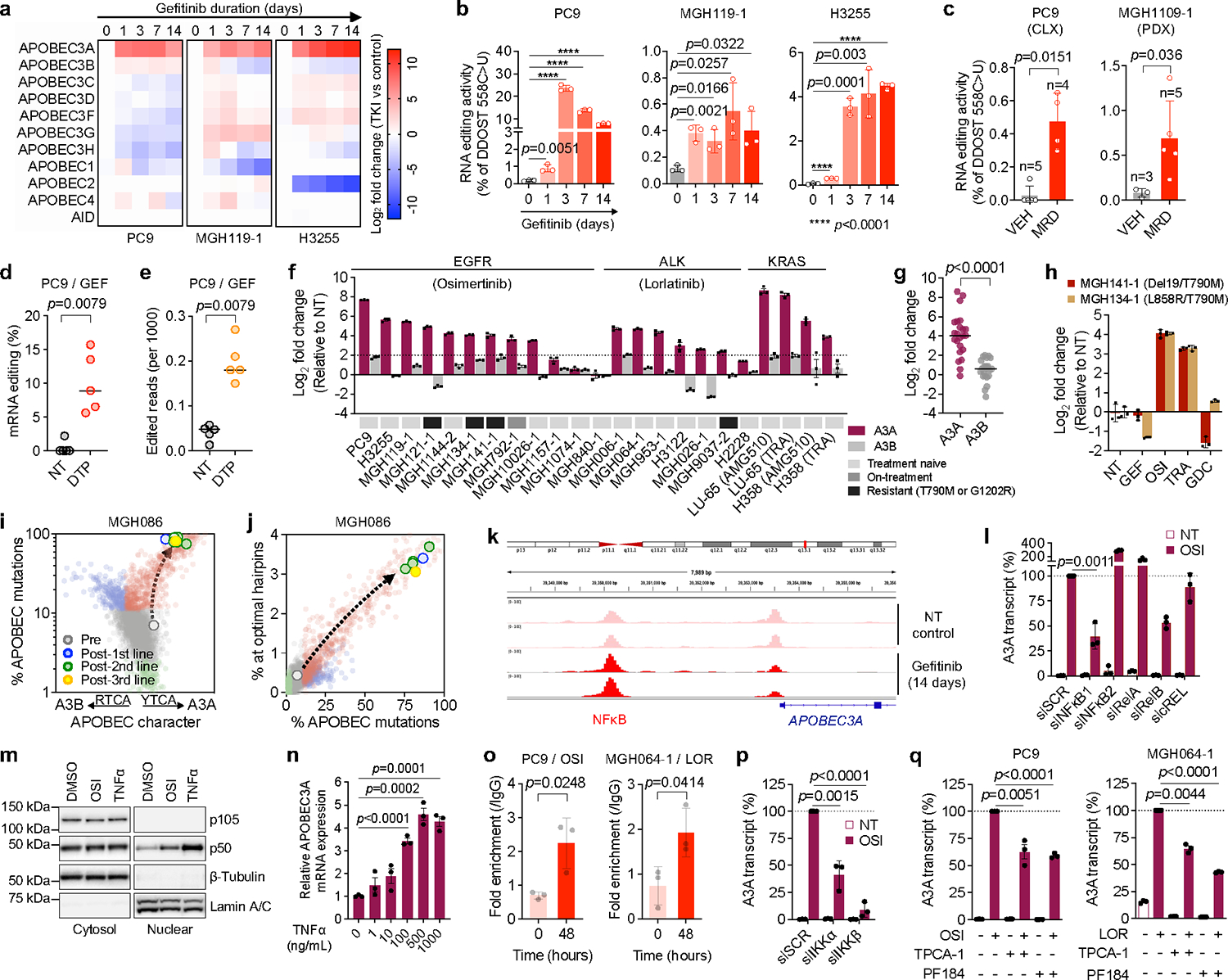
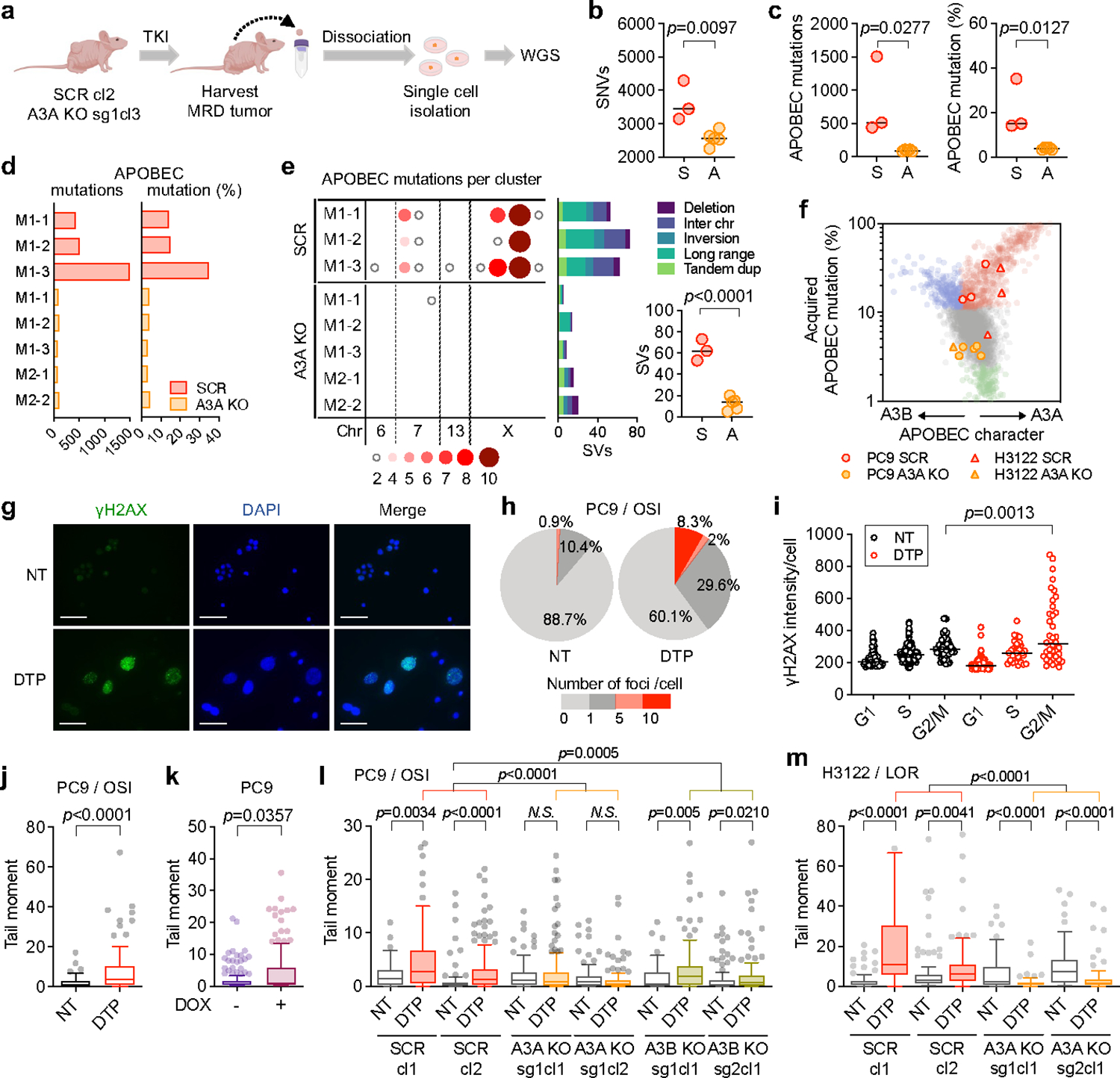
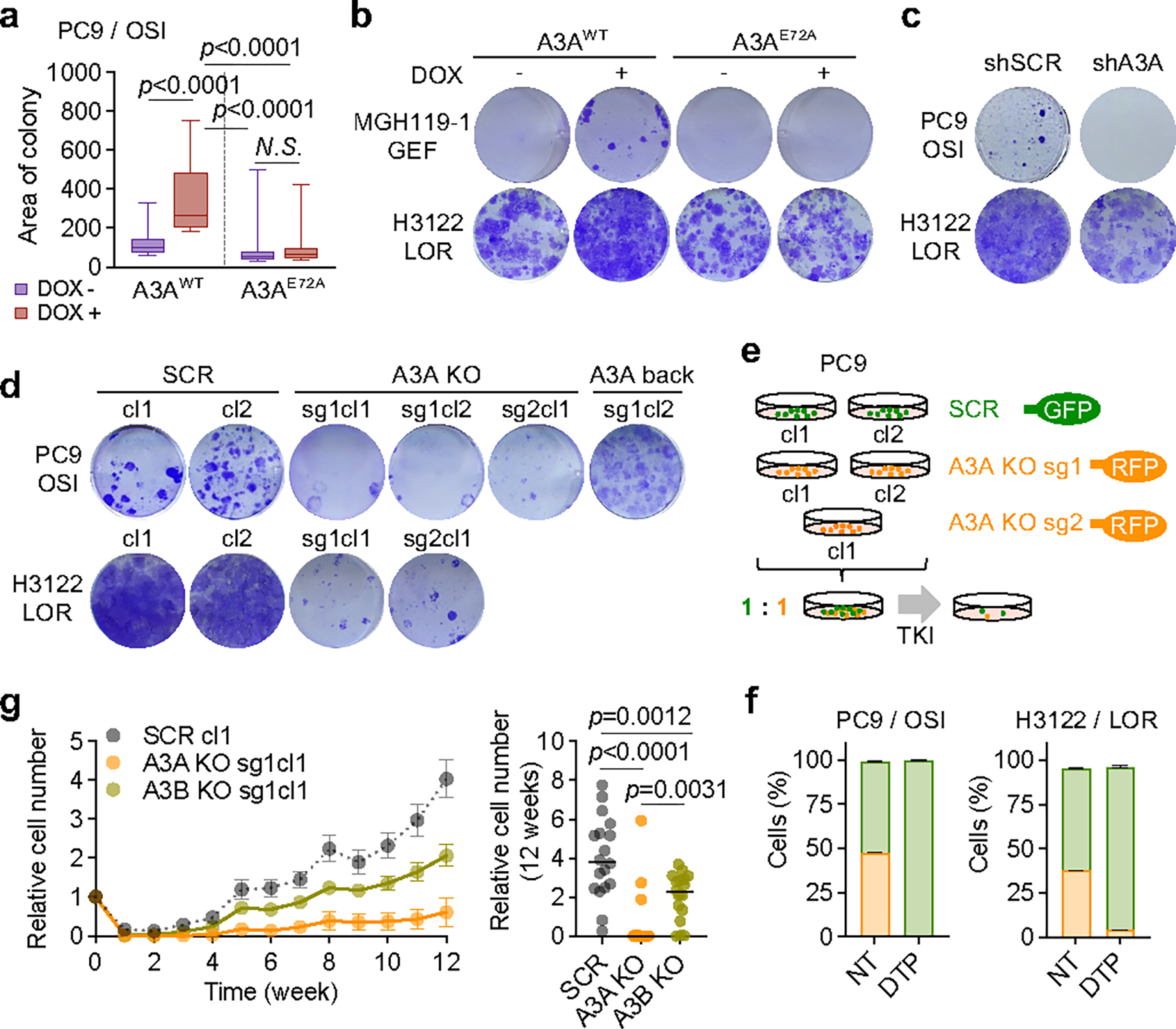
Acquired drug resistance to anticancer targeted therapies remains an unsolved clinical problem. Although many drivers of acquired drug resistance have been identified1-4, the underlying molecular mechanisms shaping tumour evolution during treatment are incompletely understood. Genomic profiling of patient tumours has implicated apolipoprotein B messenger RNA editing catalytic polypeptide-like (APOBEC) cytidine deaminases in tumour evolution; however, their role during therapy and the development of acquired drug resistance is undefined. Here we report that lung cancer targeted therapies commonly used in the clinic can induce cytidine deaminase APOBEC3A (A3A), leading to sustained mutagenesis in drug-tolerant cancer cells persisting during therapy. Therapy-induced A3A promotes the formation of double-strand DNA breaks, increasing genomic instability in drug-tolerant persisters. Deletion of A3A reduces APOBEC mutations and structural variations in persister cells and delays the development of drug resistance. APOBEC mutational signatures are enriched in tumours from patients with lung cancer who progressed after extended responses to targeted therapies. This study shows that induction of A3A in response to targeted therapies drives evolution of drug-tolerant persister cells, suggesting that suppression of A3A expression or activity may represent a potential therapeutic strategy in the prevention or delay of acquired resistance to lung cancer targeted therapy.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
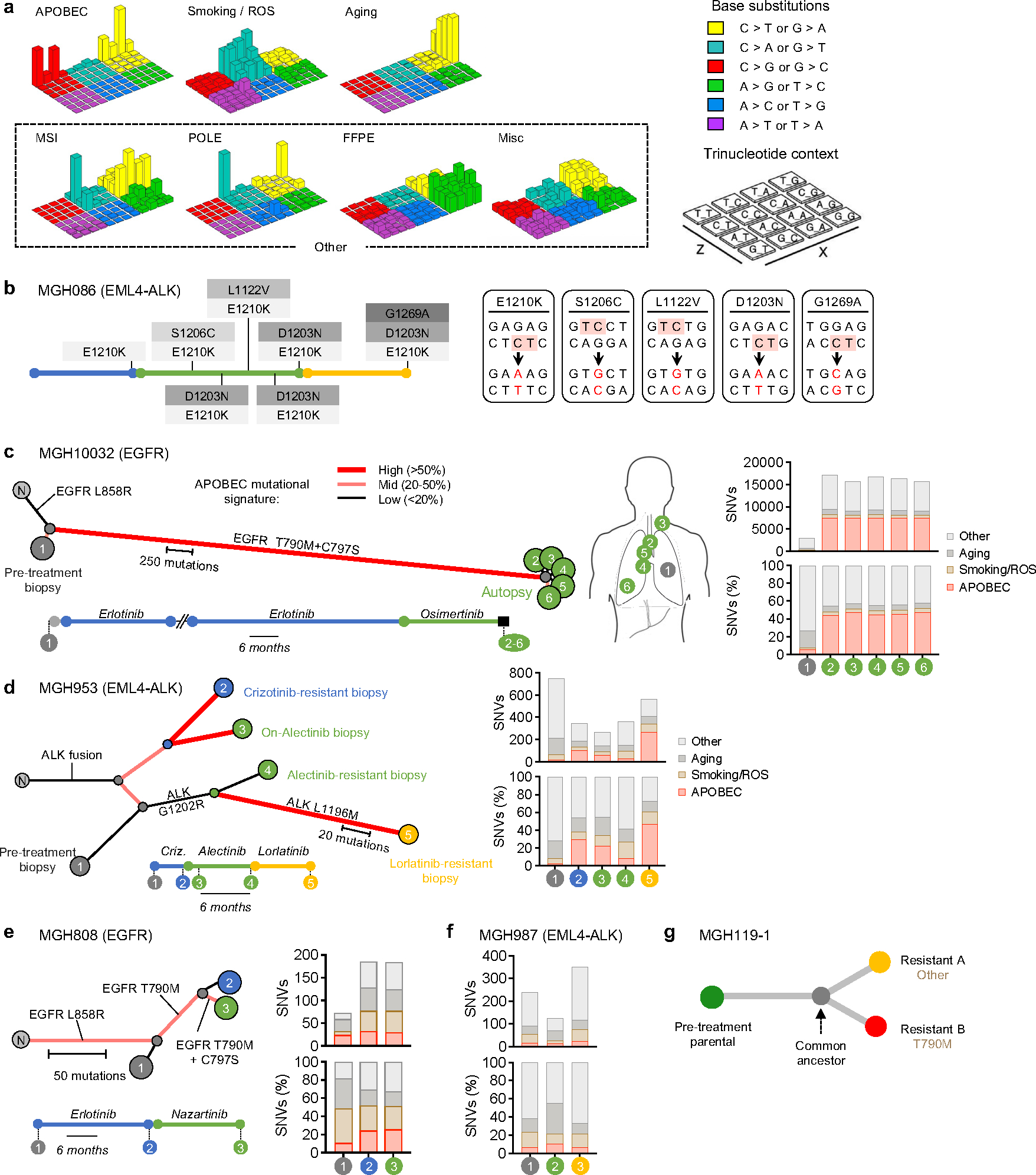
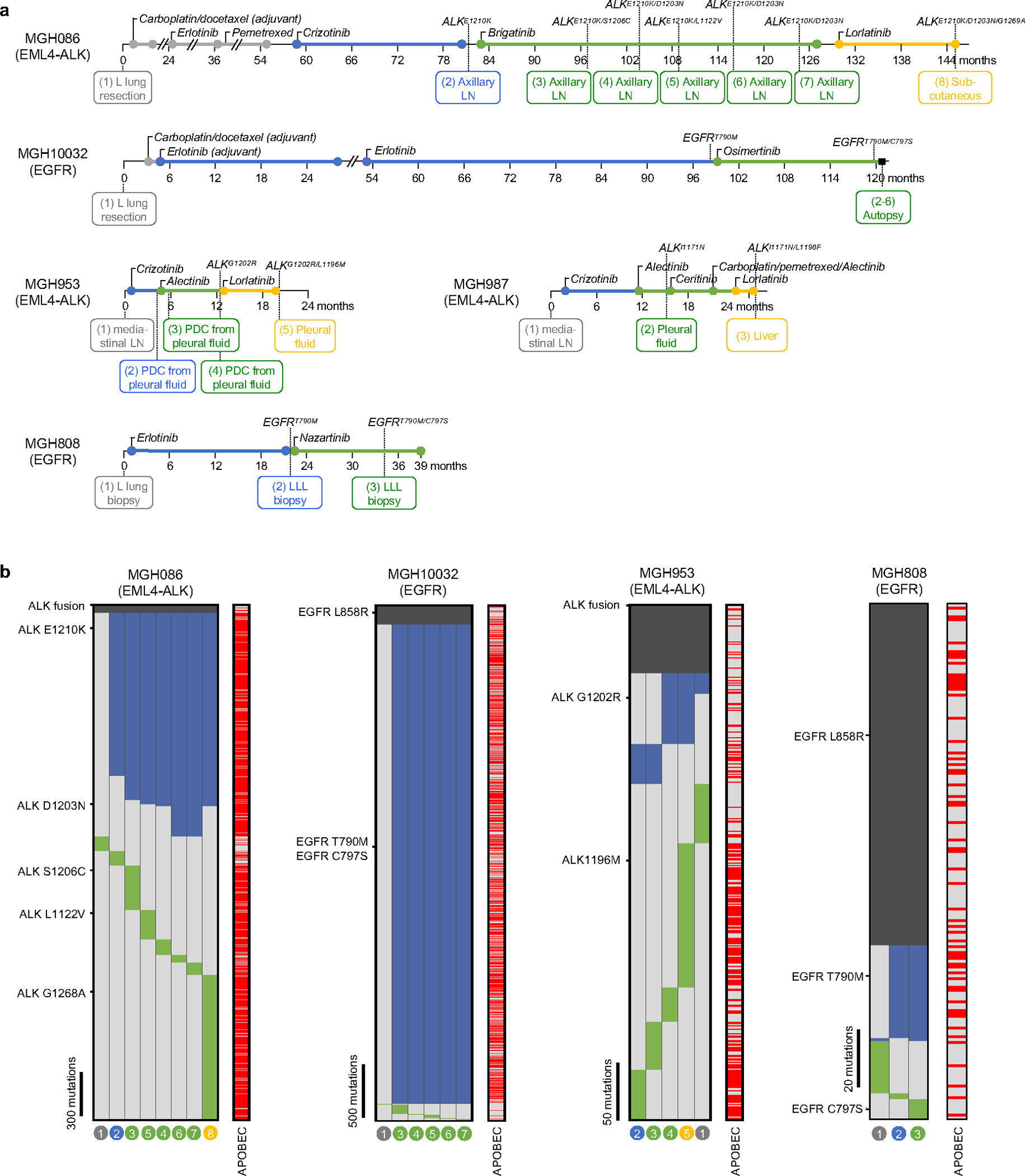
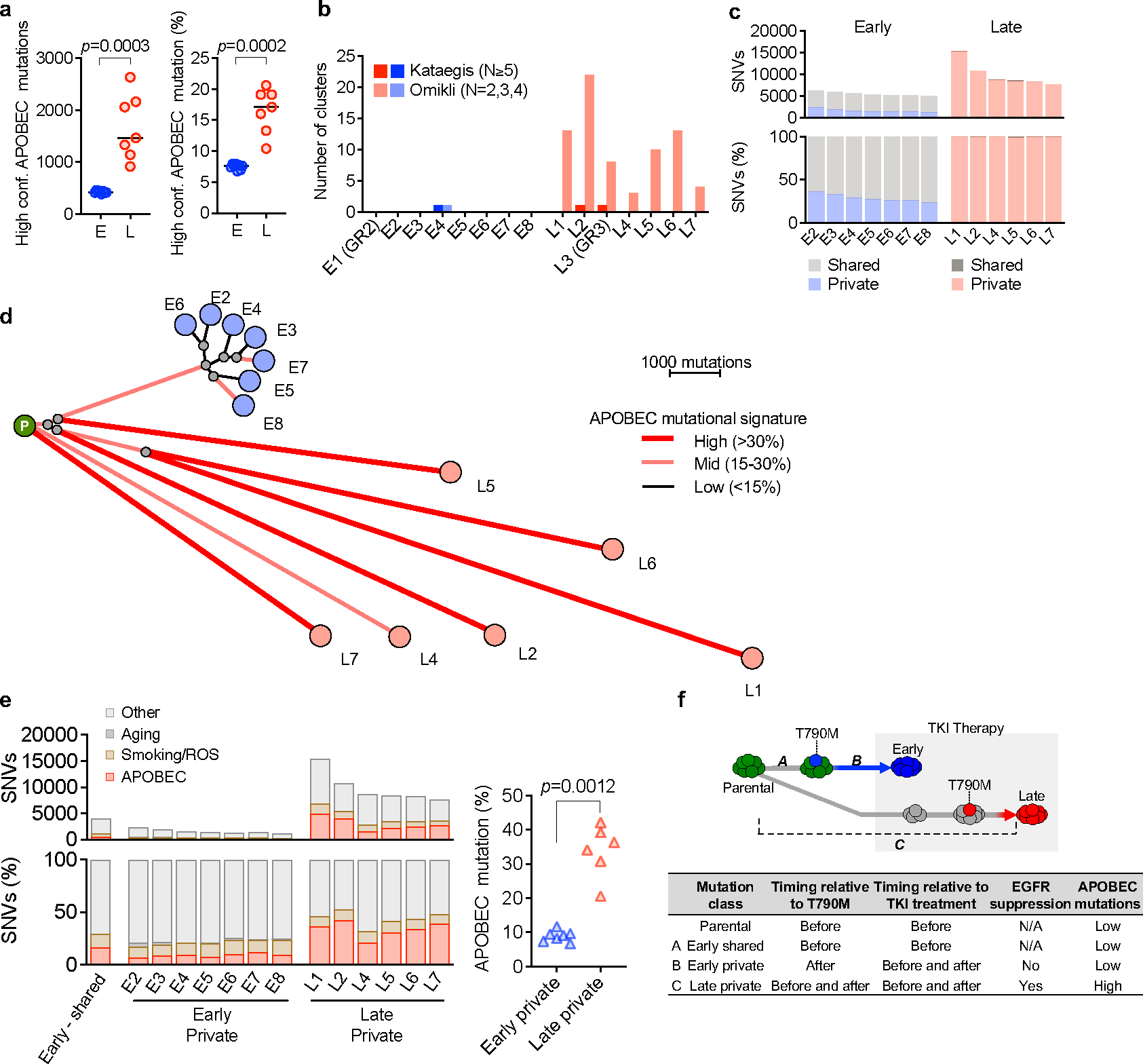
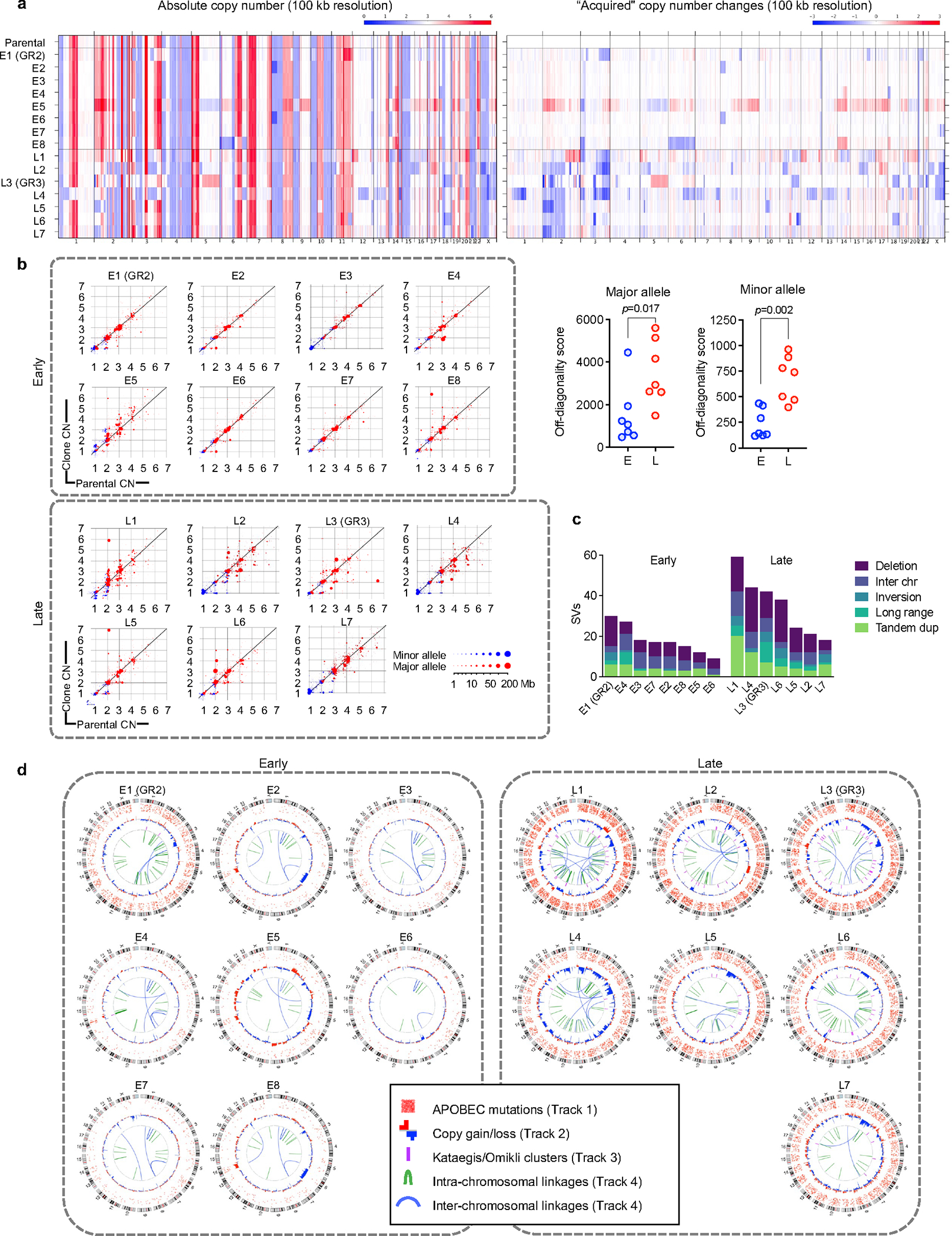
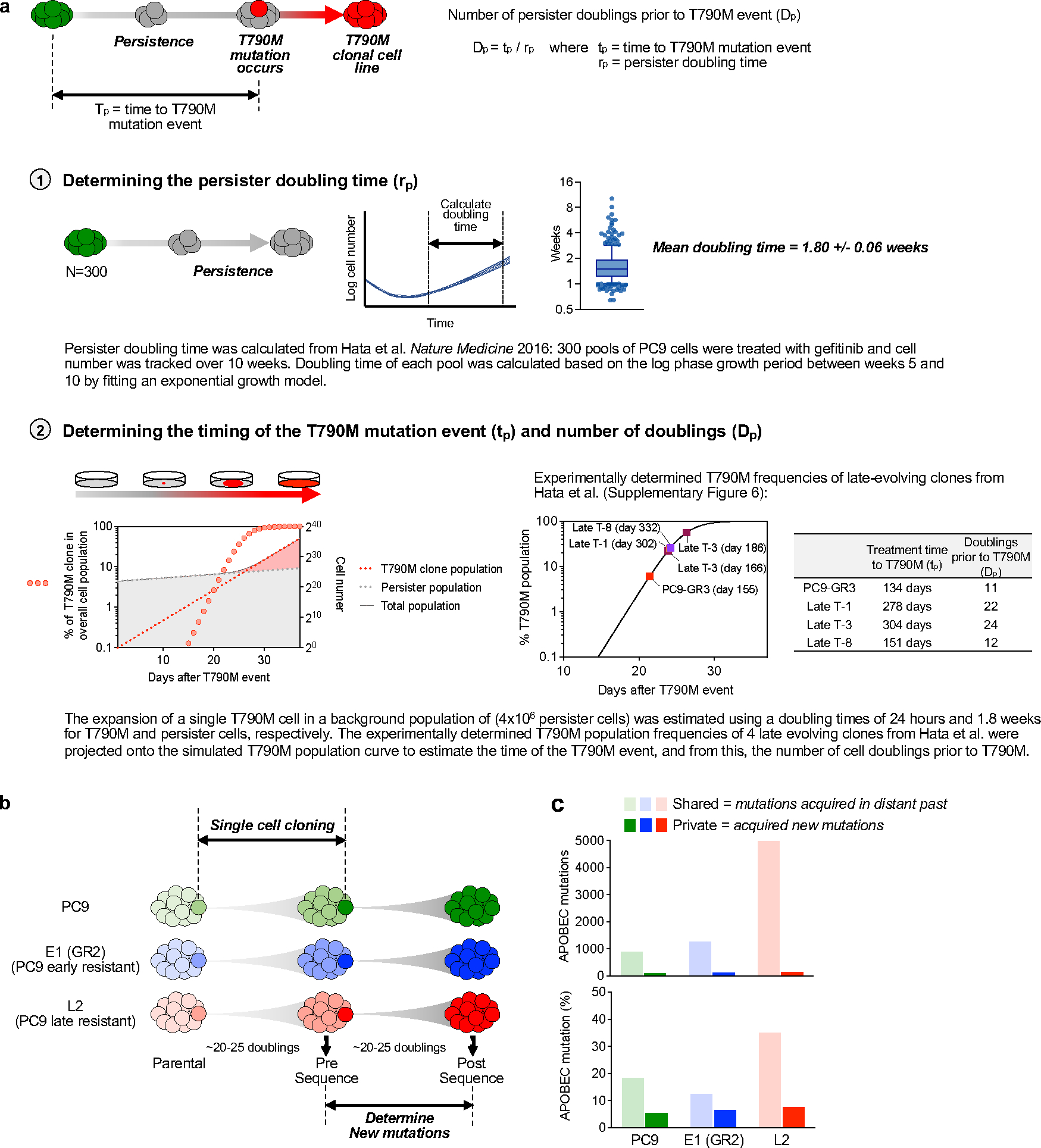
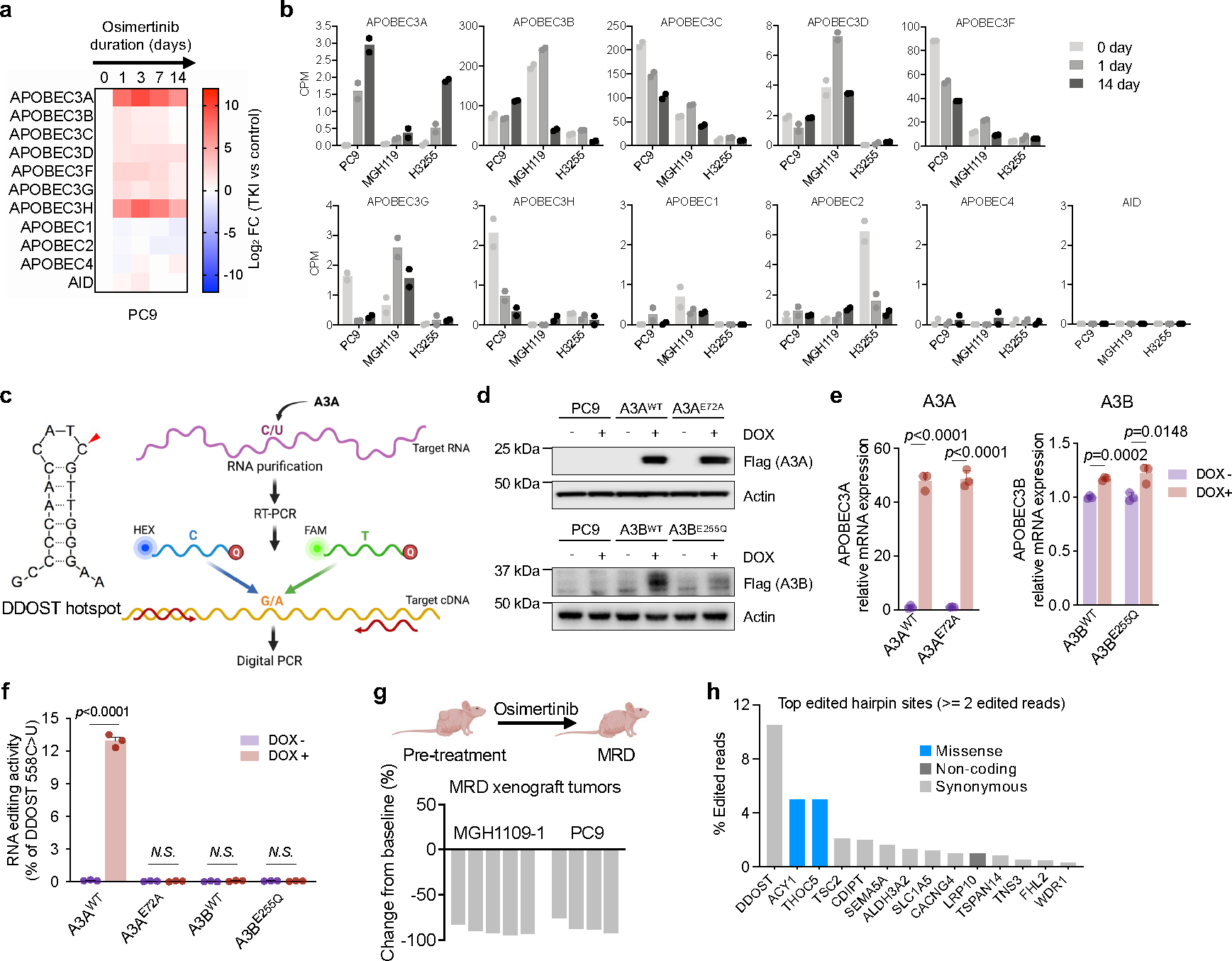
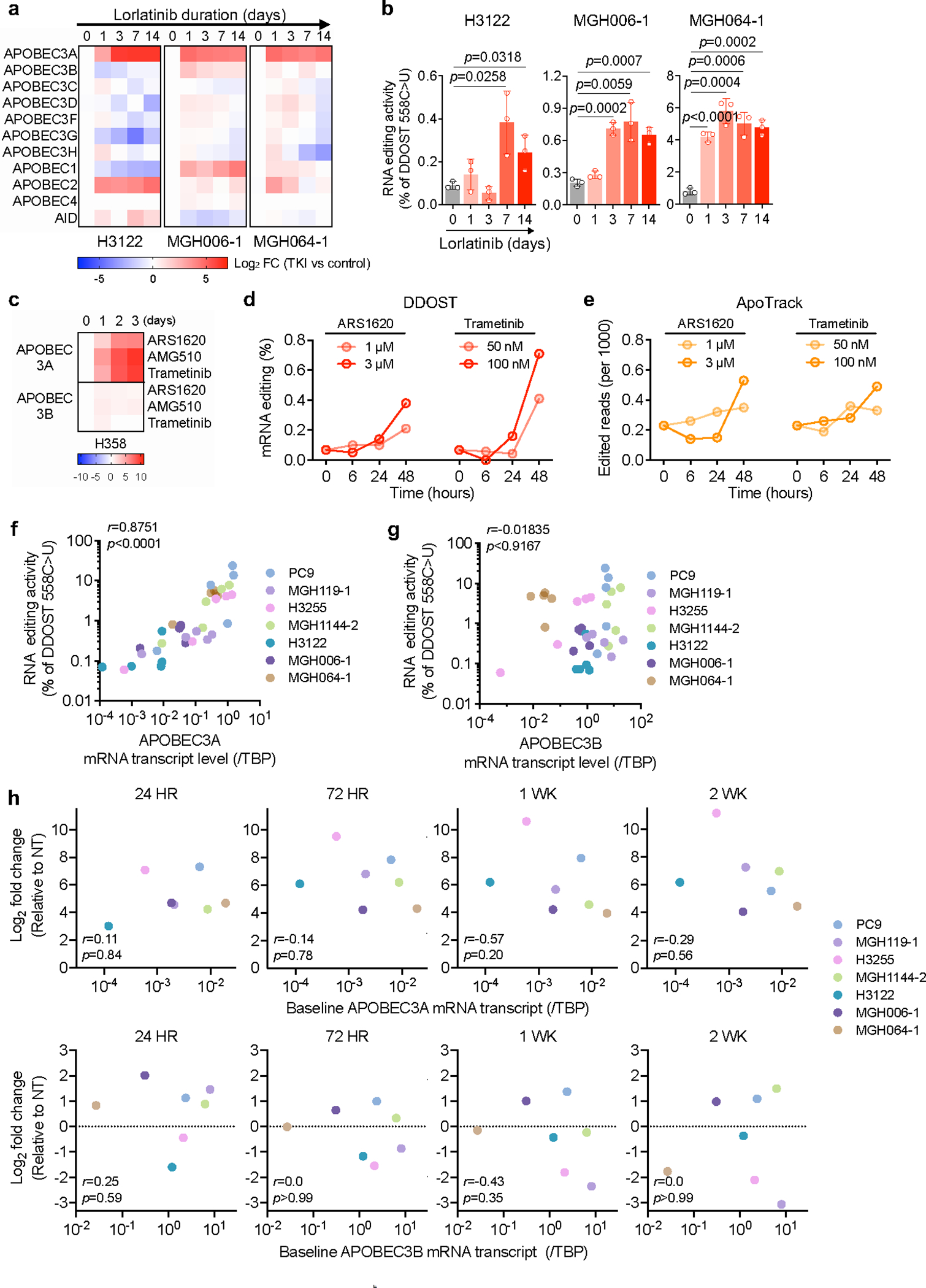
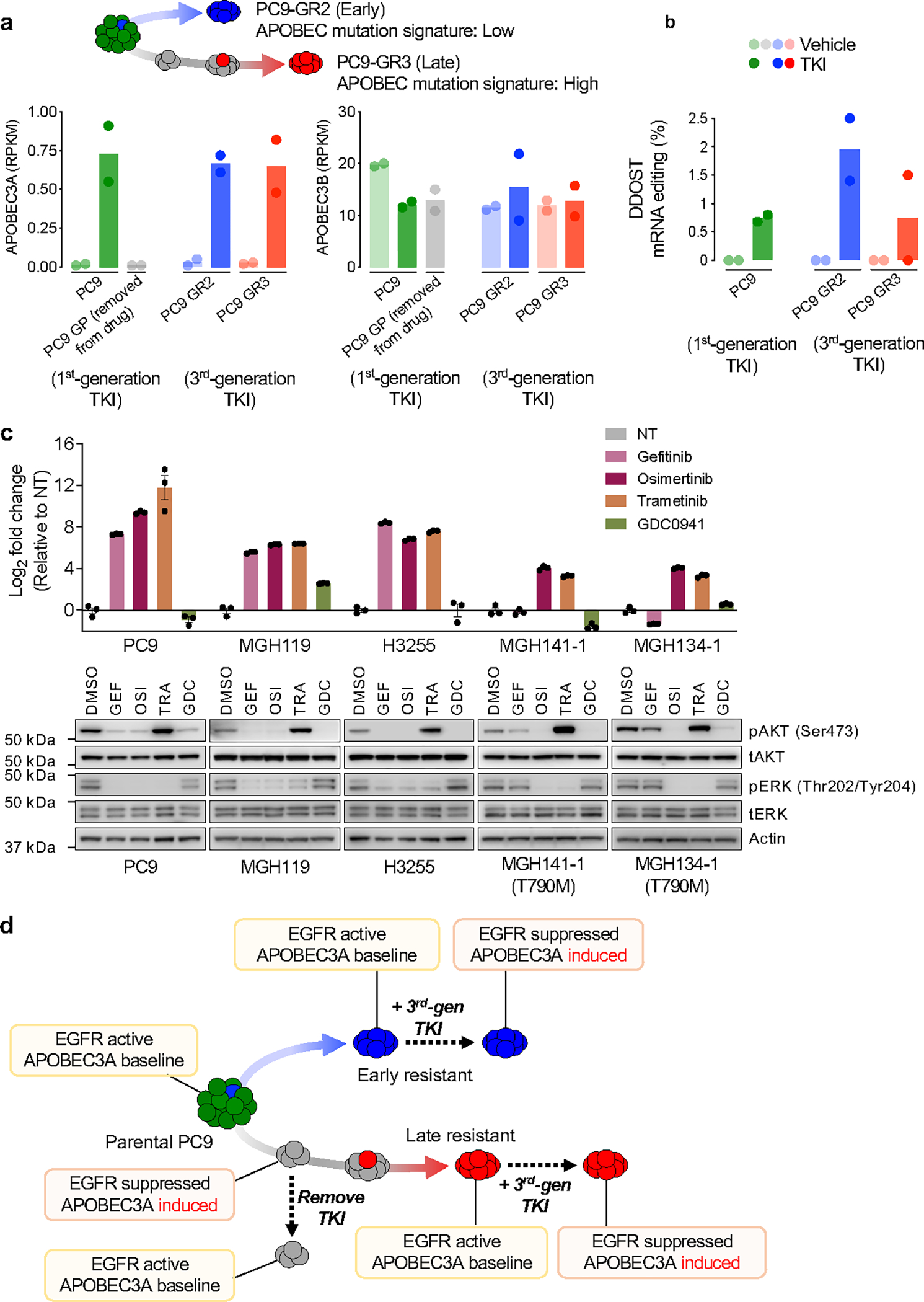
Comment in
-
APOBEC3A drives resistance.Nat Rev Drug Discov. 2023 Sep;22(9):695. doi: 10.1038/d41573-023-00122-9. Nat Rev Drug Discov. 2023. PMID: 37524811 No abstract available.
References
-
- Gainor JF et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov 6, 1118–1133 (2016). 10.1158/2159-8290.CD-16-0596 - DOI - PMC - PubMed
Extended References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
