

Regioselective aliphatic C-H functionalization using frustrated radical pairs
- PMID: 37407819
- PMCID: PMC10530363
- DOI: 10.1038/s41586-023-06131-3
Regioselective aliphatic C-H functionalization using frustrated radical pairs
Abstract
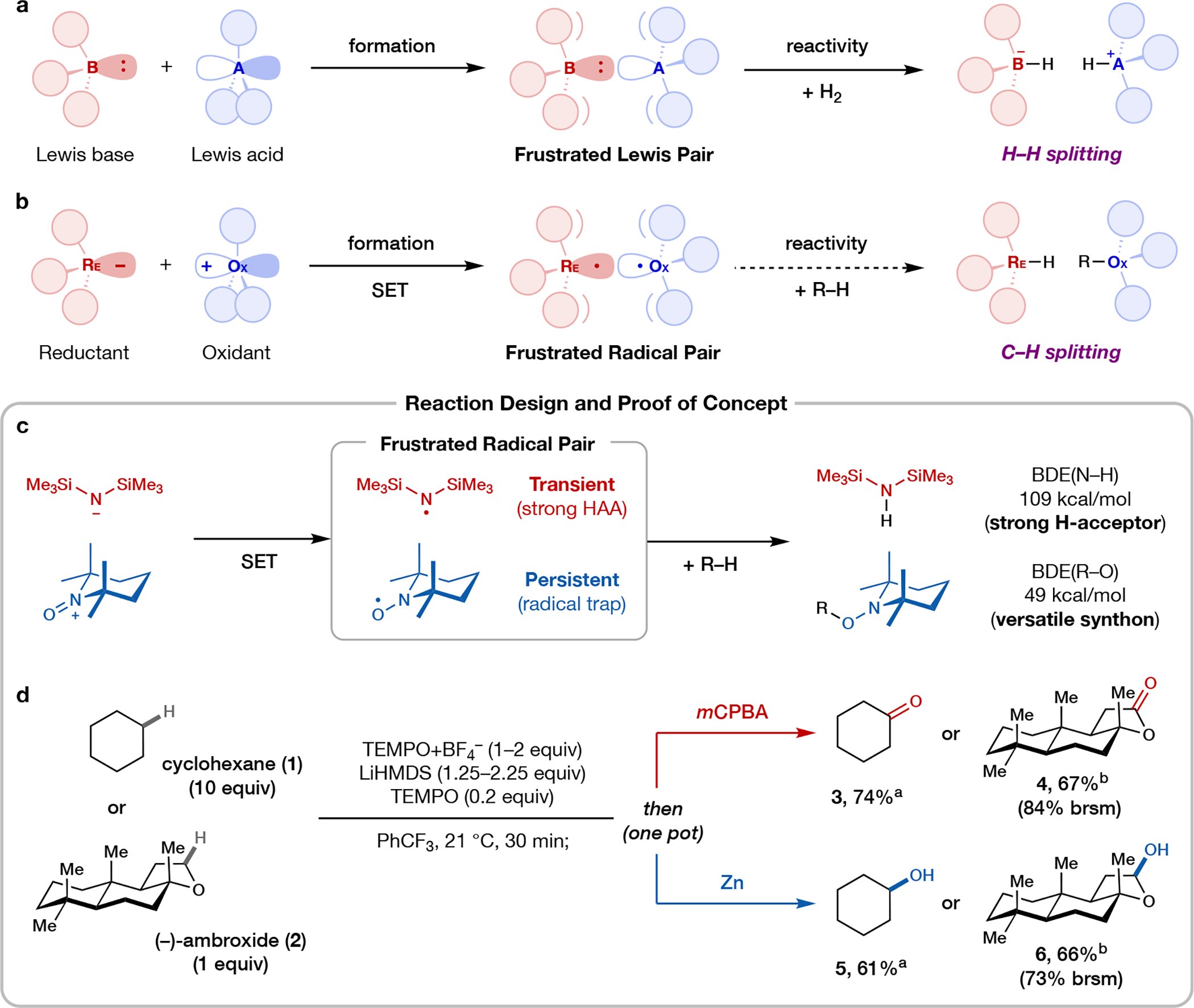
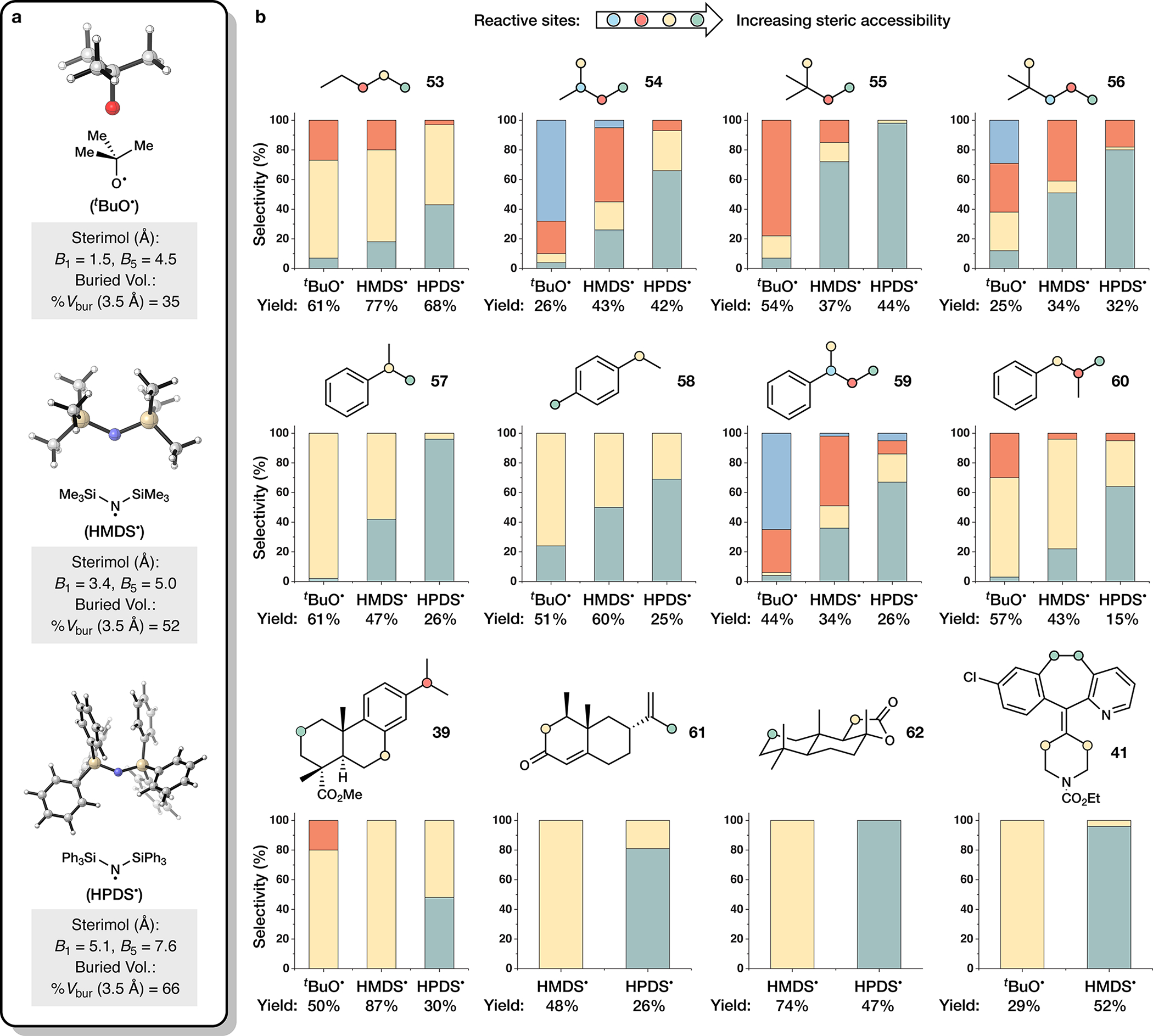
Frustrated Lewis pairs (FLPs) are well documented for the activation of small molecules such as dihydrogen and carbon dioxide1-4. Although canonical FLP chemistry is heterolytic in nature, recent work has shown that certain FLPs can undergo single-electron transfer to afford radical pairs5. Owing to steric encumbrance and/or weak bonding association, these radicals do not annihilate one another, and they have thus been named frustrated radical pairs (FRPs). Notable preliminary results suggest that FRPs may be useful reagents in chemical synthesis6-8, although their applications remain limited. Here we demonstrate that the functionalization of C(sp3)-H bonds can be accomplished using a class of FRPs generated from disilazide donors and an N-oxoammonium acceptor. Together, these species undergo single-electron transfer to generate a transient and persistent radical pair capable of cleaving unactivated C-H bonds to furnish aminoxylated products. By tuning the structure of the donor, it is possible to control regioselectivity and tailor reactivity towards tertiary, secondary or primary C-H bonds. Mechanistic studies lend strong support for the formation and involvement of radical pairs in the target reaction.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures


References
-
- Stephan DW Frustrated Lewis Pairs. J. Am. Chem. Soc. 137, 10018–10032 (2015). - PubMed
-
- Stephan DW & Erker G Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem. Int. Ed. 54, 6400–6441 (2015). - PubMed
-
- Stephan DW The broadening reach of frustrated Lewis pair chemistry. Science. 354, aaf7229 (2016). - PubMed
-
- Piers WE, Marwitz AJV & Mercier LG Mechanistic aspects of bond activation with perfluoroarylboranes. Inorg. Chem. 50, 12252–12262 (2011). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
