

To eat or not to eat mitochondria? How do host cells cope with mitophagy upon bacterial infection?
- PMID: 37410705
- PMCID: PMC10325083
- DOI: 10.1371/journal.ppat.1011471
To eat or not to eat mitochondria? How do host cells cope with mitophagy upon bacterial infection?
Abstract
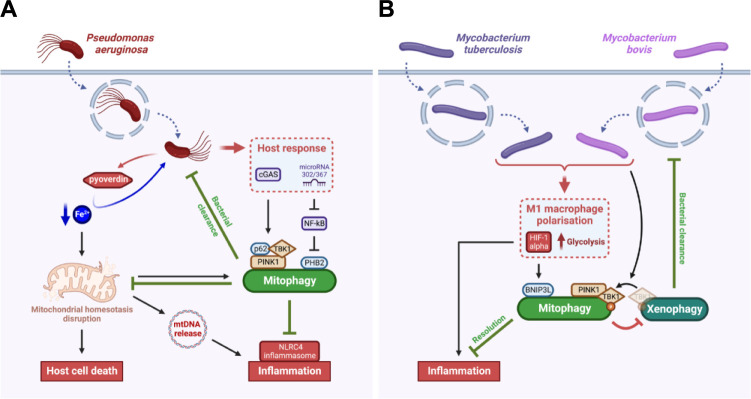
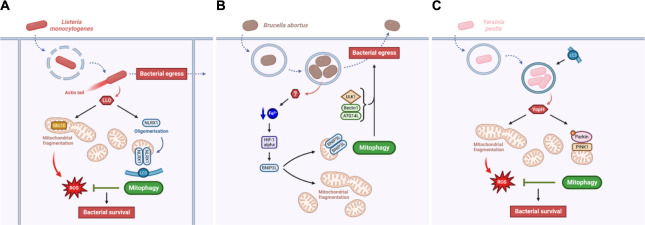
Mitochondria fulfil a plethora of cellular functions ranging from energy production to regulation of inflammation and cell death control. The fundamental role of mitochondria makes them a target of choice for invading pathogens, with either an intracellular or extracellular lifestyle. Indeed, the modulation of mitochondrial functions by several bacterial pathogens has been shown to be beneficial for bacterial survival inside their host. However, so far, relatively little is known about the importance of mitochondrial recycling and degradation pathways through mitophagy in the outcome (success or failure) of bacterial infection. On the one hand, mitophagy could be considered as a defensive response triggered by the host upon infection to maintain mitochondrial homeostasis. However, on the other hand, the pathogen itself may initiate the host mitophagy to escape from mitochondrial-mediated inflammation or antibacterial oxidative stress. In this review, we will discuss the diversity of various mechanisms of mitophagy in a general context, as well as what is currently known about the different bacterial pathogens that have developed strategies to manipulate the host mitophagy.
Copyright: © 2023 Verbeke et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



Similar articles
-
When mitophagy dictates the outcome of cellular infection: the case of Brucella abortus.Autophagy. 2023 Nov;19(11):3022-3023. doi: 10.1080/15548627.2023.2246354. Epub 2023 Aug 17. Autophagy. 2023. PMID: 37589593 Free PMC article.
-
Modulation of host mitochondrial dynamics during bacterial infection.Mitochondrion. 2020 Jul;53:140-149. doi: 10.1016/j.mito.2020.05.005. Epub 2020 May 26. Mitochondrion. 2020. PMID: 32470613 Review.
-
On the offense and defense: mitochondrial recovery programs amidst targeted pathogenic assault.FEBS J. 2022 Nov;289(22):7014-7037. doi: 10.1111/febs.16126. Epub 2021 Jul 30. FEBS J. 2022. PMID: 34270874 Free PMC article. Review.
-
Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection.BMC Biol. 2015 Apr 3;13:22. doi: 10.1186/s12915-015-0129-1. BMC Biol. 2015. PMID: 25857750 Free PMC article. Review.
-
Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy.Cell Death Differ. 2021 Mar;28(3):827-842. doi: 10.1038/s41418-020-00657-z. Epub 2020 Nov 18. Cell Death Differ. 2021. PMID: 33208889 Free PMC article. Review.
Cited by
-
Bacterial metabolism in the host and its association with virulence.Virulence. 2025 Dec;16(1):2459336. doi: 10.1080/21505594.2025.2459336. Epub 2025 Jan 31. Virulence. 2025. PMID: 39890585 Free PMC article. Review.
-
SARS-CoV-2 Orphan Gene ORF10 Contributes to More Severe COVID-19 Disease.medRxiv [Preprint]. 2023 Nov 27:2023.11.27.23298847. doi: 10.1101/2023.11.27.23298847. medRxiv. 2023. PMID: 38076862 Free PMC article. Preprint.
-
Inhibition of Unc-51-like-kinase is mitoprotective during Pseudomonas aeruginosa infection in corneal epithelial cells.mSphere. 2025 Feb 25;10(2):e0053724. doi: 10.1128/msphere.00537-24. Epub 2025 Jan 10. mSphere. 2025. PMID: 39791872 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
