

Oncogene-like addiction to aneuploidy in human cancers
- PMID: 37410869
- PMCID: PMC10753973
- DOI: 10.1126/science.adg4521
Oncogene-like addiction to aneuploidy in human cancers
Abstract
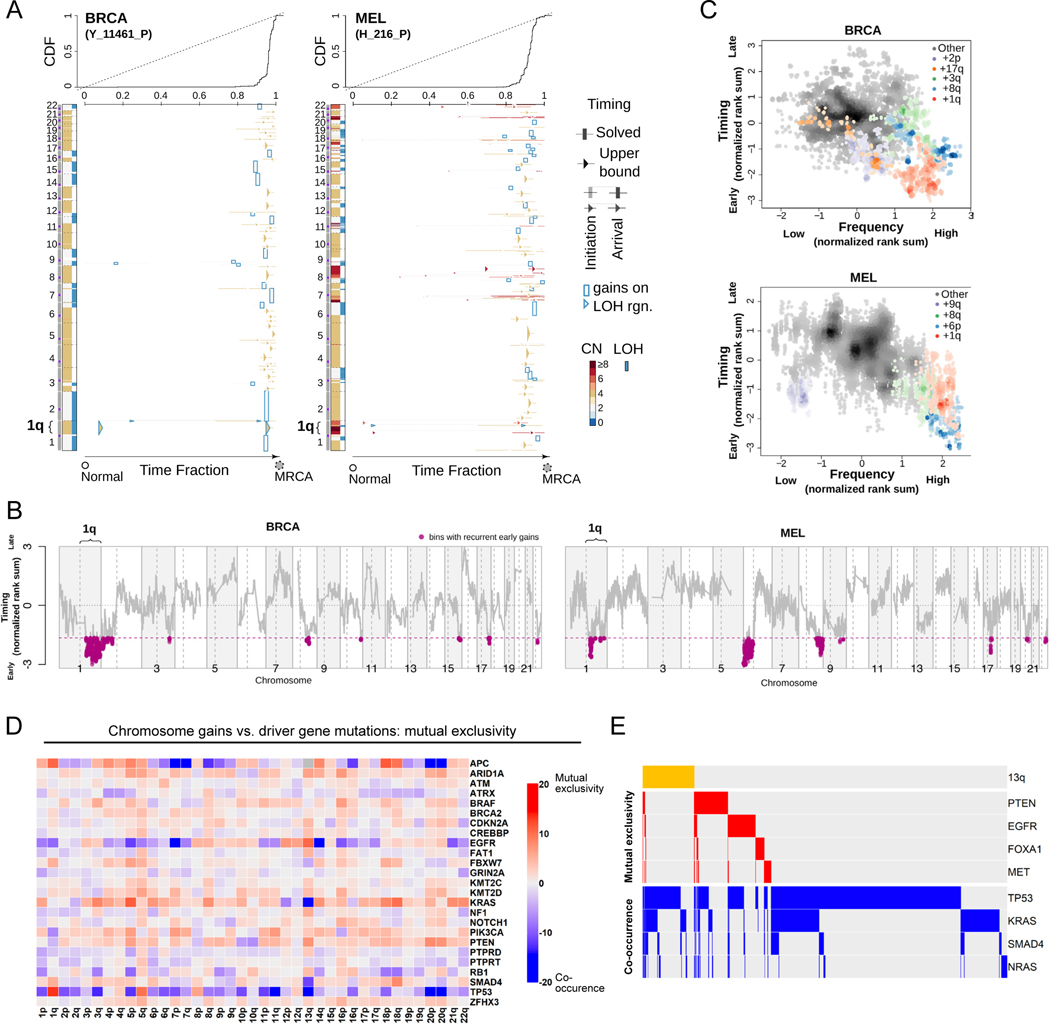
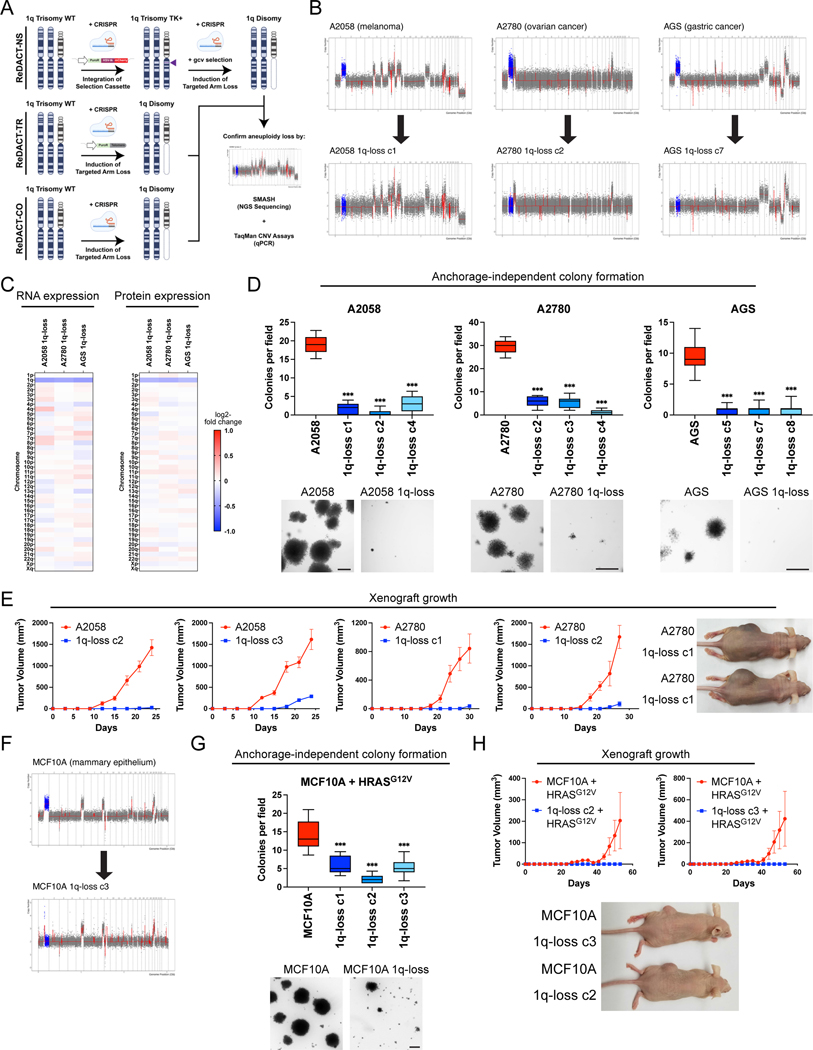
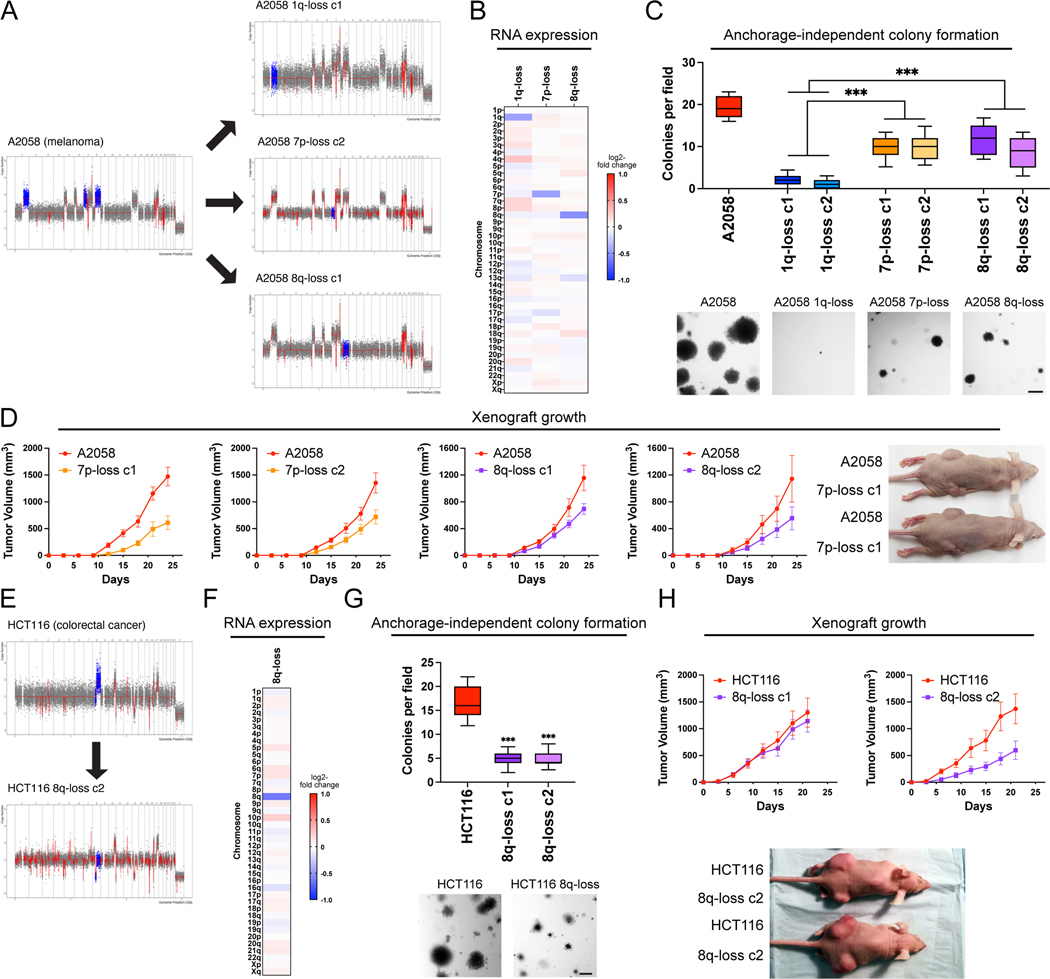
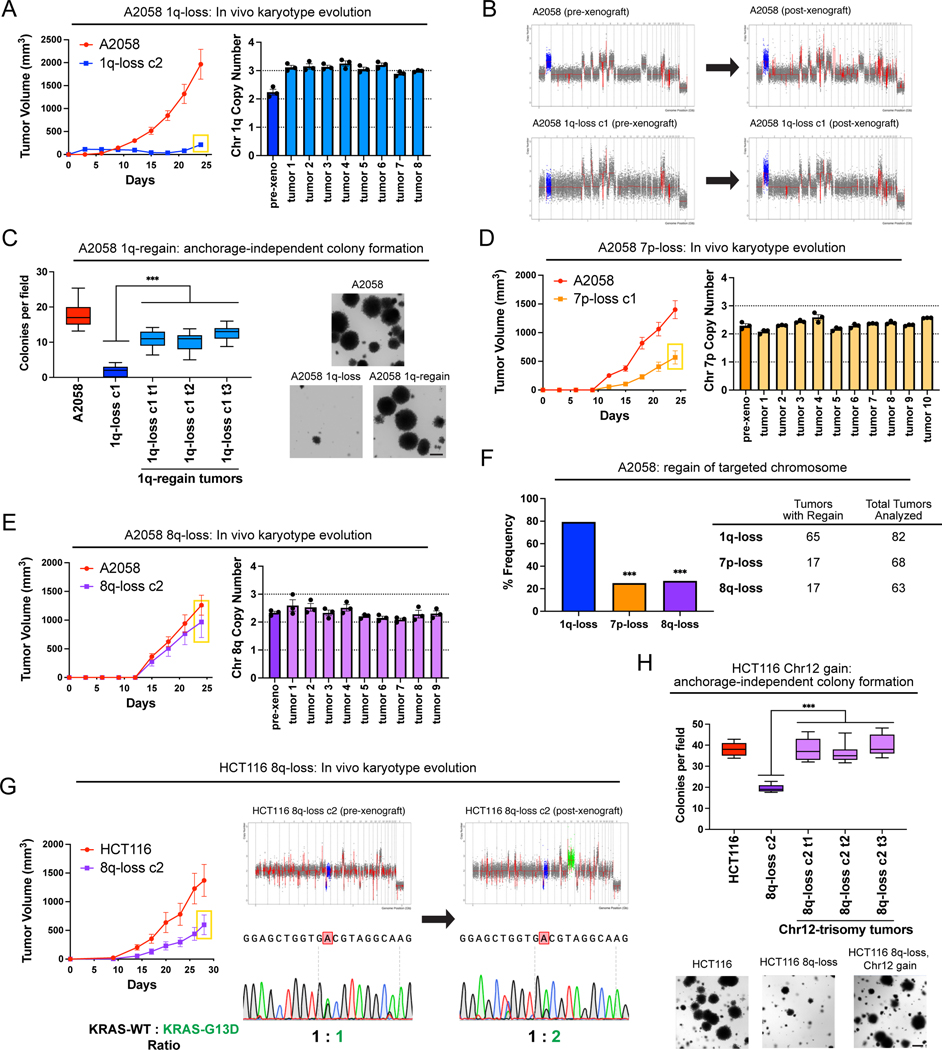
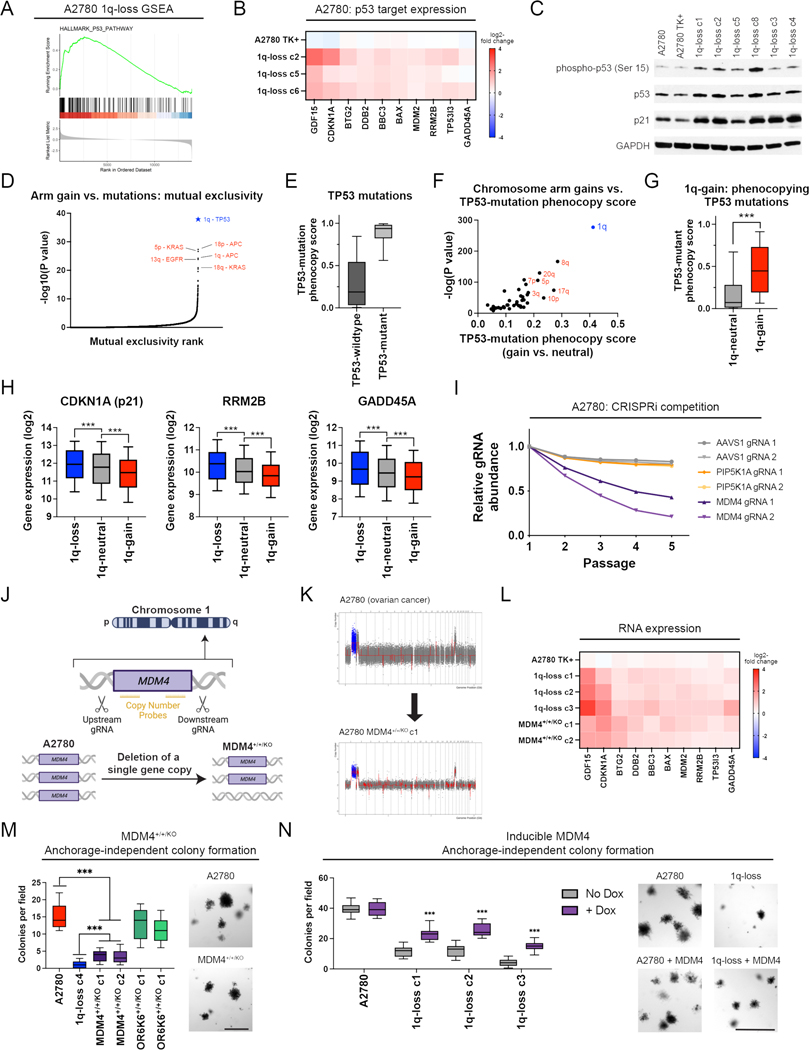
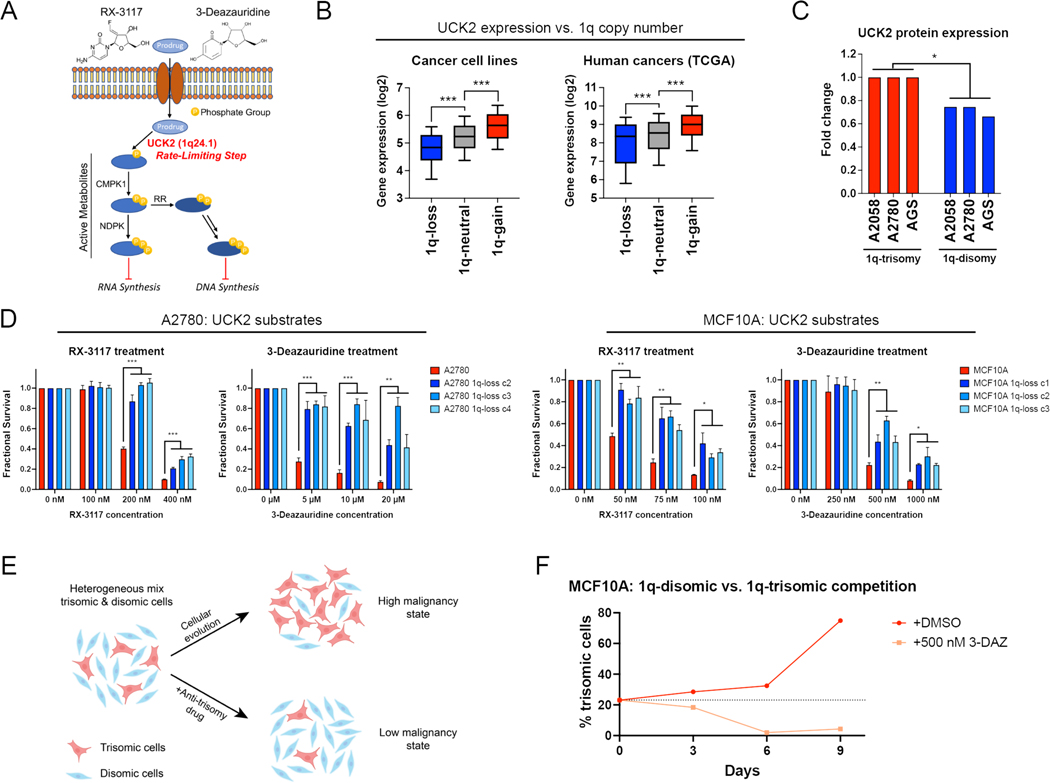
Most cancers exhibit aneuploidy, but its functional significance in tumor development is controversial. Here, we describe ReDACT (Restoring Disomy in Aneuploid cells using CRISPR Targeting), a set of chromosome engineering tools that allow us to eliminate specific aneuploidies from cancer genomes. Using ReDACT, we created a panel of isogenic cells that have or lack common aneuploidies, and we demonstrate that trisomy of chromosome 1q is required for malignant growth in cancers harboring this alteration. Mechanistically, gaining chromosome 1q increases the expression of MDM4 and suppresses p53 signaling, and we show that TP53 mutations are mutually exclusive with 1q aneuploidy in human cancers. Thus, tumor cells can be dependent on specific aneuploidies, raising the possibility that these "aneuploidy addictions" could be targeted as a therapeutic strategy.
Conflict of interest statement
Figures
Update of
-
Oncogene-like addiction to aneuploidy in human cancers.bioRxiv [Preprint]. 2023 Jan 10:2023.01.09.523344. doi: 10.1101/2023.01.09.523344. bioRxiv. 2023. Update in: Science. 2023 Aug 25;381(6660):eadg4521. doi: 10.1126/science.adg4521. PMID: 36711674 Free PMC article. Updated. Preprint.
Comment in
-
Human Tumor Cells Display an Oncogene-like Addiction to Aneuploidy.Cancer Discov. 2023 Sep 6;13(9):1956. doi: 10.1158/2159-8290.CD-RW2023-110. Cancer Discov. 2023. PMID: 37449722
References
-
- Gordon DJ, Resio B, Pellman D, Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet 13, 189–203 (2012). - PubMed
-
- Boveri T, Concerning the Origin of Malignant Tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci 121, 1–84 (2008). - PubMed
-
- Hardy PA, Zacharias H, Reappraisal of the Hansemann-Boveri hypothesis on the origin of tumors. Cell Biol. Int 29, 983–992 (2005). - PubMed
-
- Vasudevan A, Schukken KM, Sausville EL, Girish V, Adebambo OA, Sheltzer JM, Aneuploidy as a promoter and suppressor of malignant growth. Nat. Rev. Cancer 21, 89–103 (2021). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
