

Monoacylglycerol Lipase Protects the Presynaptic Cannabinoid 1 Receptor from Desensitization by Endocannabinoids after Persistent Inflammation
- PMID: 37414560
- PMCID: PMC10376933
- DOI: 10.1523/JNEUROSCI.0037-23.2023
Monoacylglycerol Lipase Protects the Presynaptic Cannabinoid 1 Receptor from Desensitization by Endocannabinoids after Persistent Inflammation
Abstract
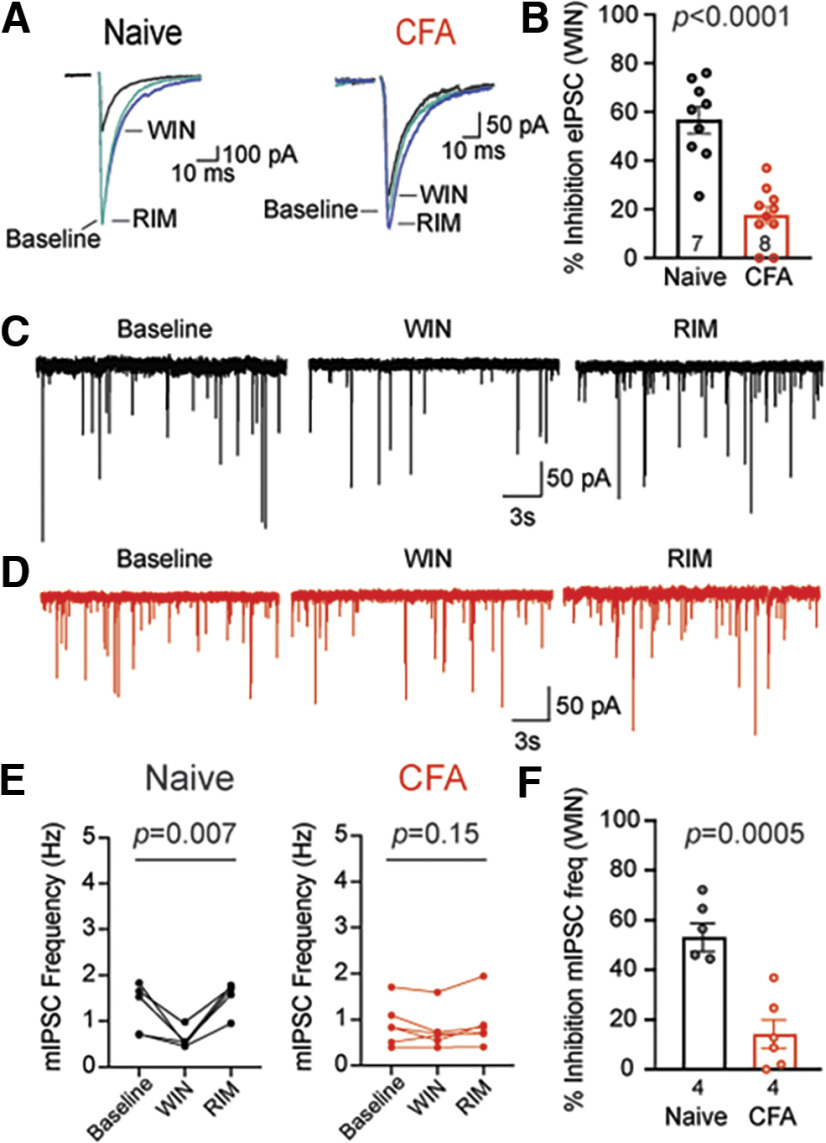
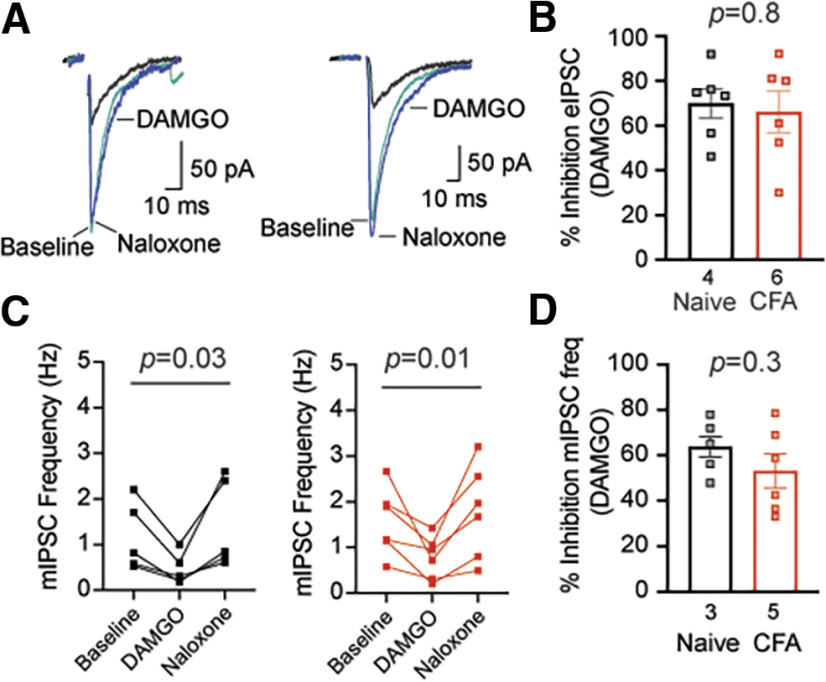
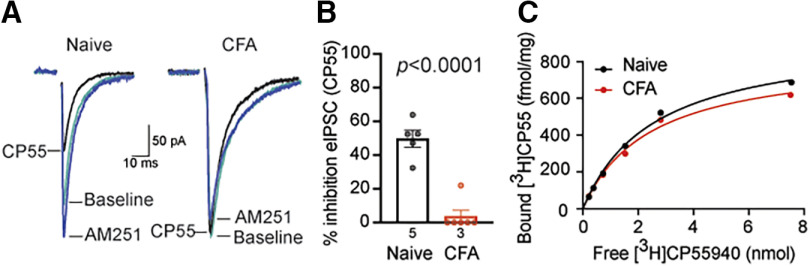
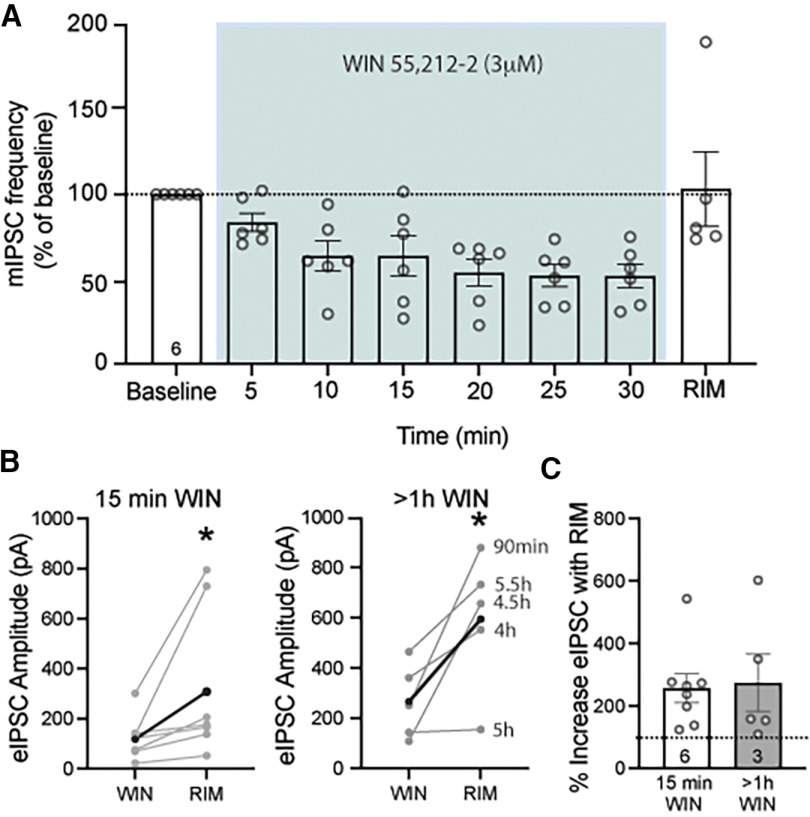
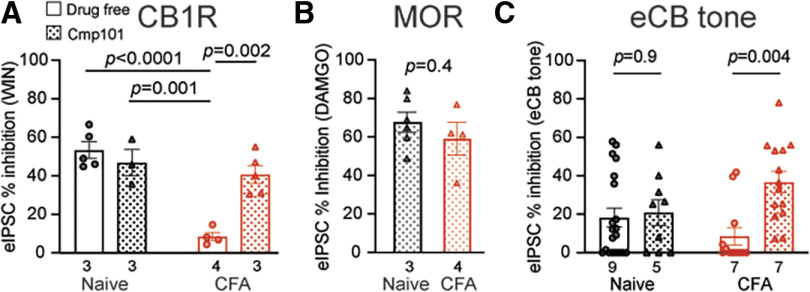
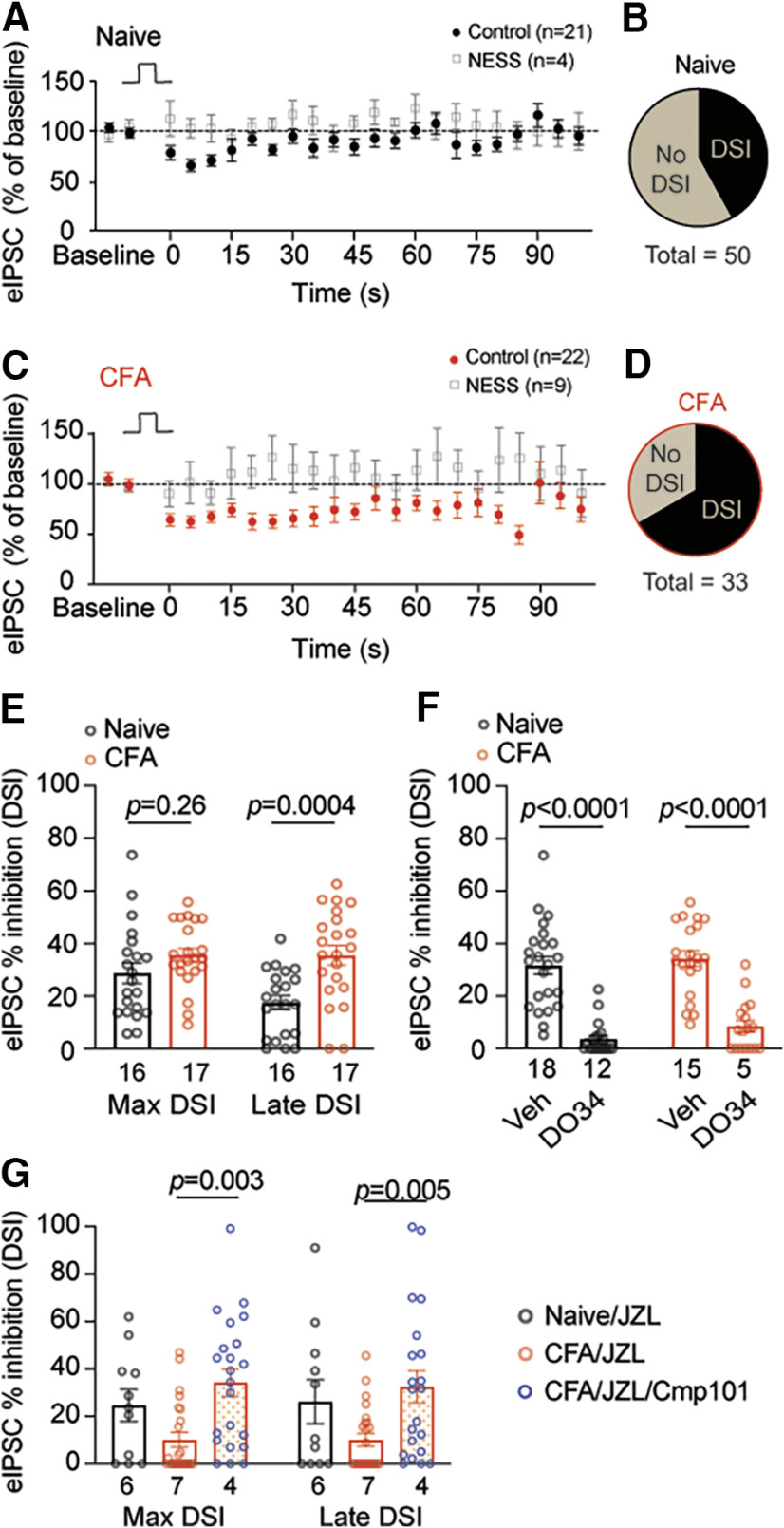
Cannabinoid-targeted pain therapies are increasing with the expansion of cannabis legalization, however, their efficacy may be limited by pain-induced adaptations in the cannabinoid system. Cannabinoid receptor subtype 1 (CB1R) inhibition of spontaneous, GABAergic miniature IPSCs (mIPSCs) and evoked IPSCs (eIPSCs) in the ventrolateral periaqueductal gray (vlPAG) were compared in slices from naive and inflamed male and female Sprague Dawley rats. Complete Freund's Adjuvant (CFA) injections into the hindpaw induced persistent inflammation. In naive rats, exogenous cannabinoid agonists robustly reduce both eIPSCs and mIPSCs. After 5-7 d of inflammation, the effects of exogenous cannabinoids are significantly reduced because of CB1R desensitization via GRK2/3, as function is recovered in the presence of the GRK2/3 inhibitor, Compound 101 (Cmp101). Inhibition of GABA release by presynaptic μ-opioid receptors in the vlPAG does not desensitize with persistent inflammation. Unexpectedly, while CB1R desensitization significantly reduces the inhibition produced by exogenous agonists, depolarization-induced suppression of inhibition protocols that promote 2-arachidonoylglycerol (2-AG) synthesis exhibit prolonged CB1R activation after inflammation. 2-AG tone is detected in slices from CFA-treated rats when GRK2/3 is blocked, suggesting an increase in 2-AG synthesis after persistent inflammation. Inhibiting 2-AG degradation with the monoacylglycerol lipase (MAGL) inhibitor JZL184 during inflammation results in the desensitization of CB1Rs by endocannabinoids that is reversed with Cmp101. Collectively, these data indicate that persistent inflammation primes CB1Rs for desensitization, and MAGL degradation of 2-AG protects CB1Rs from desensitization in inflamed rats. These adaptations with inflammation have important implications for the development of cannabinoid-based pain therapeutics targeting MAGL and CB1Rs.SIGNIFICANCE STATEMENT Presynaptic G-protein-coupled receptors are resistant to desensitization. Here we find that persistent inflammation increases endocannabinoid levels, priming presynaptic cannabinoid 1 receptors for desensitization on subsequent addition of exogenous agonists. Despite the reduced efficacy of exogenous agonists, endocannabinoids have prolonged efficacy after persistent inflammation. Endocannabinoids readily induce cannabinoid 1 receptor desensitization if their degradation is blocked, indicating that endocannabinoid concentrations are maintained at subdesensitizing levels and that degradation is critical for maintaining endocannabinoid regulation of presynaptic GABA release in the ventrolateral periaqueductal gray during inflammatory states. These adaptations with inflammation have important implications for the development of cannabinoid-based pain therapies.
Keywords: GPCR; desensitization; endocannabinoid; inflammation; periaqueductal gray; presynaptic.
Copyright © 2023 the authors.
Figures
Similar articles
-
Compensatory Activation of Cannabinoid CB2 Receptor Inhibition of GABA Release in the Rostral Ventromedial Medulla in Inflammatory Pain.J Neurosci. 2017 Jan 18;37(3):626-636. doi: 10.1523/JNEUROSCI.1310-16.2016. J Neurosci. 2017. PMID: 28100744 Free PMC article.
-
Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus.J Neurosci. 2007 Jan 31;27(5):1211-9. doi: 10.1523/JNEUROSCI.4159-06.2007. J Neurosci. 2007. PMID: 17267577 Free PMC article.
-
Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment.Pharmacol Res. 2013 Jan;67(1):94-109. doi: 10.1016/j.phrs.2012.10.013. Epub 2012 Nov 2. Pharmacol Res. 2013. PMID: 23127915 Free PMC article.
-
The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors.Acta Physiol (Oxf). 2012 Feb;204(2):267-76. doi: 10.1111/j.1748-1716.2011.02280.x. Epub 2011 Apr 22. Acta Physiol (Oxf). 2012. PMID: 21418147 Free PMC article. Review.
-
Targeting the cannabinoid system for pain relief?Acta Anaesthesiol Taiwan. 2013 Dec;51(4):161-70. doi: 10.1016/j.aat.2013.10.004. Epub 2013 Dec 25. Acta Anaesthesiol Taiwan. 2013. PMID: 24529672 Review.
Cited by
-
Key differences in regulation of opioid receptors localized to presynaptic terminals compared to somas: Relevance for novel therapeutics.Neuropharmacology. 2023 Mar 15;226:109408. doi: 10.1016/j.neuropharm.2022.109408. Epub 2022 Dec 28. Neuropharmacology. 2023. PMID: 36584882 Free PMC article. Review.
-
Endocannabinoids regulate enteric neuron-glia networks and visceral hypersensitivity following inflammation through a glial-dependent mechanism.Glia. 2024 Nov;72(11):2095-2114. doi: 10.1002/glia.24599. Epub 2024 Aug 12. Glia. 2024. PMID: 39132860
References
-
- Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF (2009) Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol 16:411–420. - PMC - PubMed
-
- Ahn K, Smith SE, Liimatta MB, Beidler D, Sadagopan N, Dudley DT, Young T, Wren P, Zhang Y, Swaney S, Van Becelaere K, Blankman JL, Nomura DK, Bhattachar SN, Stiff C, Nomanbhoy TK, Weerapana E, Johnson DS, Cravatt BF (2011) Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther 338:114–124. - PMC - PubMed
-
- Anderson WB, Gould MJ, Torres RD, Mitchell VA, Vaughan CW (2014) Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine inflammatory pain model. Neuropharmacology 81:224–230. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials