

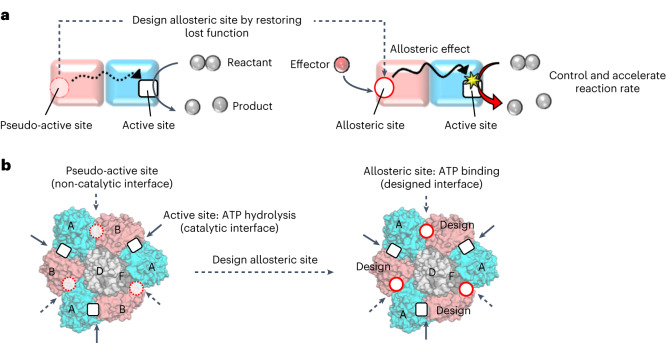
Design of allosteric sites into rotary motor V1-ATPase by restoring lost function of pseudo-active sites
- PMID: 37414880
- PMCID: PMC10624635
- DOI: 10.1038/s41557-023-01256-4
Design of allosteric sites into rotary motor V1-ATPase by restoring lost function of pseudo-active sites
Abstract
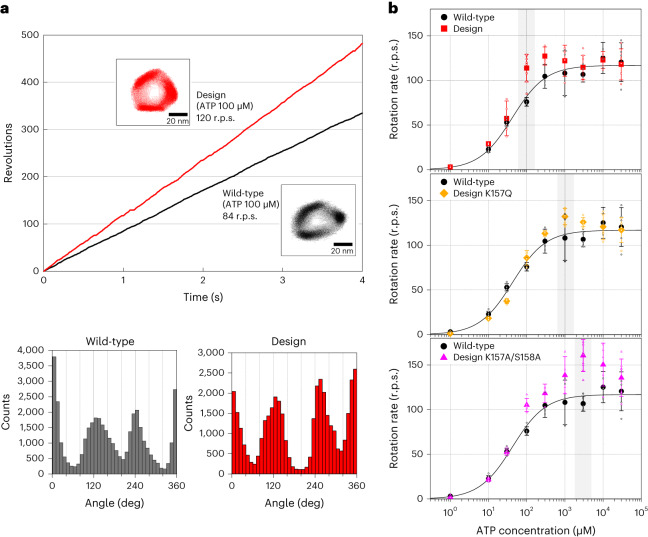
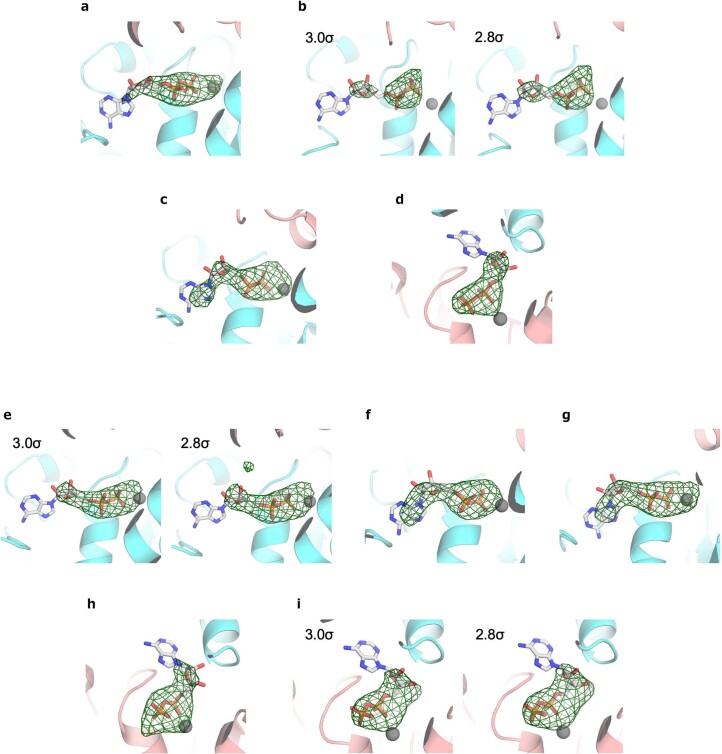
Allostery produces concerted functions of protein complexes by orchestrating the cooperative work between the constituent subunits. Here we describe an approach to create artificial allosteric sites in protein complexes. Certain protein complexes contain subunits with pseudo-active sites, which are believed to have lost functions during evolution. Our hypothesis is that allosteric sites in such protein complexes can be created by restoring the lost functions of pseudo-active sites. We used computational design to restore the lost ATP-binding ability of the pseudo-active site in the B subunit of a rotary molecular motor, V1-ATPase. Single-molecule experiments with X-ray crystallography analyses revealed that binding of ATP to the designed allosteric site boosts this V1's activity compared with the wild-type, and the rotation rate can be tuned by modulating ATP's binding affinity. Pseudo-active sites are widespread in nature, and our approach shows promise as a means of programming allosteric control over concerted functions of protein complexes.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
Structure and dynamics of rotary V1 motor.Cell Mol Life Sci. 2018 May;75(10):1789-1802. doi: 10.1007/s00018-018-2758-3. Epub 2018 Jan 31. Cell Mol Life Sci. 2018. PMID: 29387903 Free PMC article. Review.
-
Inter-subunit interaction and quaternary rearrangement defined by the central stalk of prokaryotic V1-ATPase.EMBO Rep. 2009 Nov;10(11):1228-34. doi: 10.1038/embor.2009.202. Epub 2009 Sep 25. EMBO Rep. 2009. PMID: 19779483 Free PMC article.
-
Basic properties of rotary dynamics of the molecular motor Enterococcus hirae V1-ATPase.J Biol Chem. 2013 Nov 8;288(45):32700-32707. doi: 10.1074/jbc.M113.506329. Epub 2013 Oct 2. J Biol Chem. 2013. PMID: 24089518 Free PMC article.
-
Molecular structure and rotary dynamics of Enterococcus hirae V₁-ATPase.IUBMB Life. 2014 Sep;66(9):624-30. doi: 10.1002/iub.1311. Epub 2014 Sep 17. IUBMB Life. 2014. PMID: 25229752 Review.
-
The presence of the alternatively spliced A2 cassette in the vacuolar H+-ATPase subunit A prevents assembly of the V1 catalytic domain.Eur J Biochem. 1999 Nov;266(1):293-301. doi: 10.1046/j.1432-1327.1999.00874.x. Eur J Biochem. 1999. PMID: 10542077
Cited by
-
Uncovering the design principles of supramolecular assemblies through manipulation of the structures, dynamics, and functions.Biophys Physicobiol. 2022 Aug 27;19:e190031. doi: 10.2142/biophysico.bppb-v19.0031. eCollection 2022. Biophys Physicobiol. 2022. PMID: 36349329 Free PMC article. No abstract available.
-
Modeling reveals the strength of weak interactions in stacked-ring assembly.Biophys J. 2024 Jul 2;123(13):1763-1780. doi: 10.1016/j.bpj.2024.05.015. Epub 2024 May 18. Biophys J. 2024. PMID: 38762753 Free PMC article.
-
Allo-targeting of the kinase domain: Insights from in silico studies and comparison with experiments.Curr Opin Struct Biol. 2024 Feb;84:102770. doi: 10.1016/j.sbi.2023.102770. Epub 2024 Jan 11. Curr Opin Struct Biol. 2024. PMID: 38211377 Free PMC article. Review.
-
Structure-based engineering of Tor complexes reveals that two types of yeast TORC1 produce distinct phenotypes.J Cell Sci. 2024 Feb 15;137(4):jcs261625. doi: 10.1242/jcs.261625. Epub 2024 Feb 28. J Cell Sci. 2024. PMID: 38415789 Free PMC article.
-
De novo design of ATPase based on a blueprint optimized for harboring the P-loop motif.Protein Sci. 2025 Jun;34(6):e70132. doi: 10.1002/pro.70132. Protein Sci. 2025. PMID: 40364444 Free PMC article.
References
-
- Wang, J. et al. Ligand-triggered allosteric ADP release primes a plant NLR complex. Science364, eaav5868 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 18H05420/MEXT | Japan Society for the Promotion of Science (JSPS)
- 18H05424/MEXT | Japan Society for the Promotion of Science (JSPS)
- JPMJPR20E6/MEXT | JST | Precursory Research for Embryonic Science and Technology (PRESTO)
- JPMJPR13AD/MEXT | JST | Precursory Research for Embryonic Science and Technology (PRESTO)
- BINDS0471/Japan Agency for Medical Research and Development (AMED)
LinkOut - more resources
Full Text Sources
