

Genetic and pharmacological inhibition of GRPR protects against acute kidney injury via attenuating renal inflammation and necroptosis
- PMID: 37415332
- PMCID: PMC10492025
- DOI: 10.1016/j.ymthe.2023.06.016
Genetic and pharmacological inhibition of GRPR protects against acute kidney injury via attenuating renal inflammation and necroptosis
Abstract
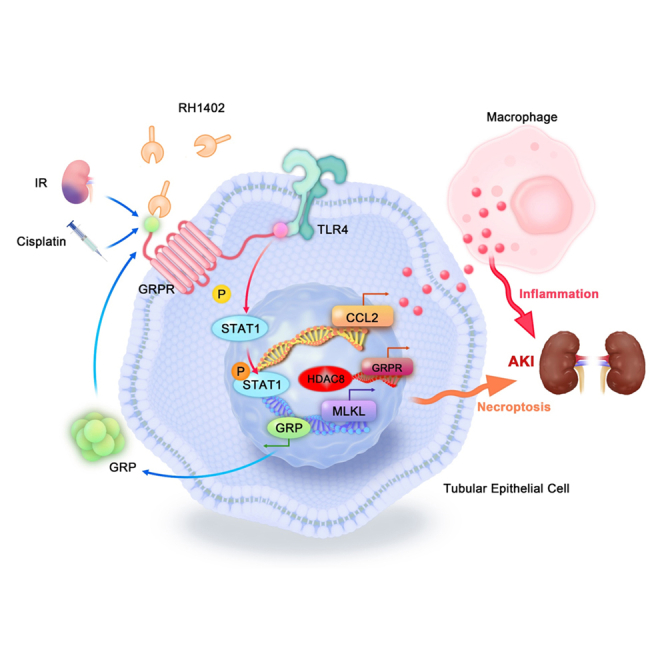
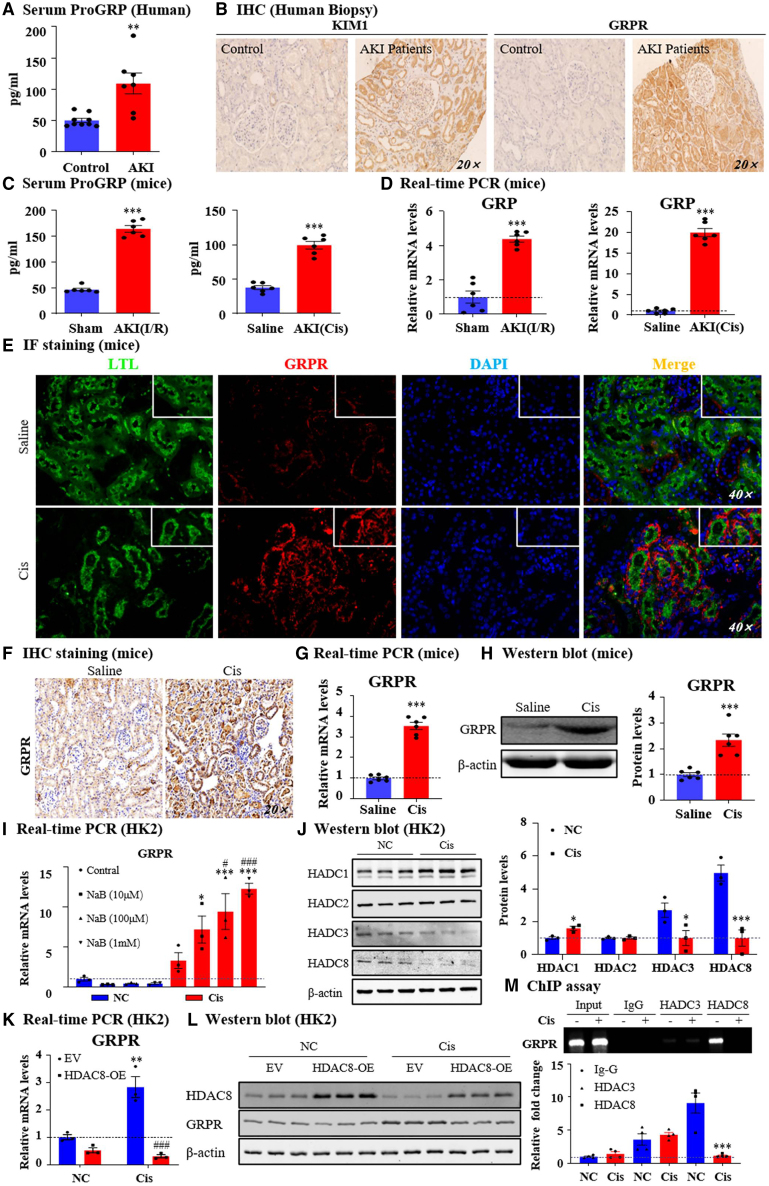
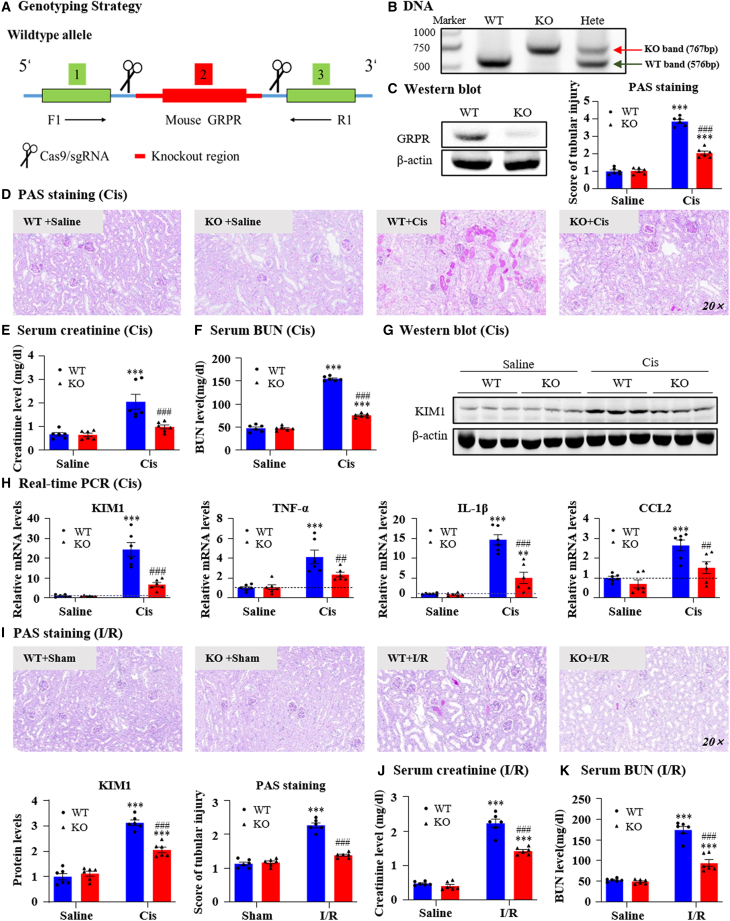
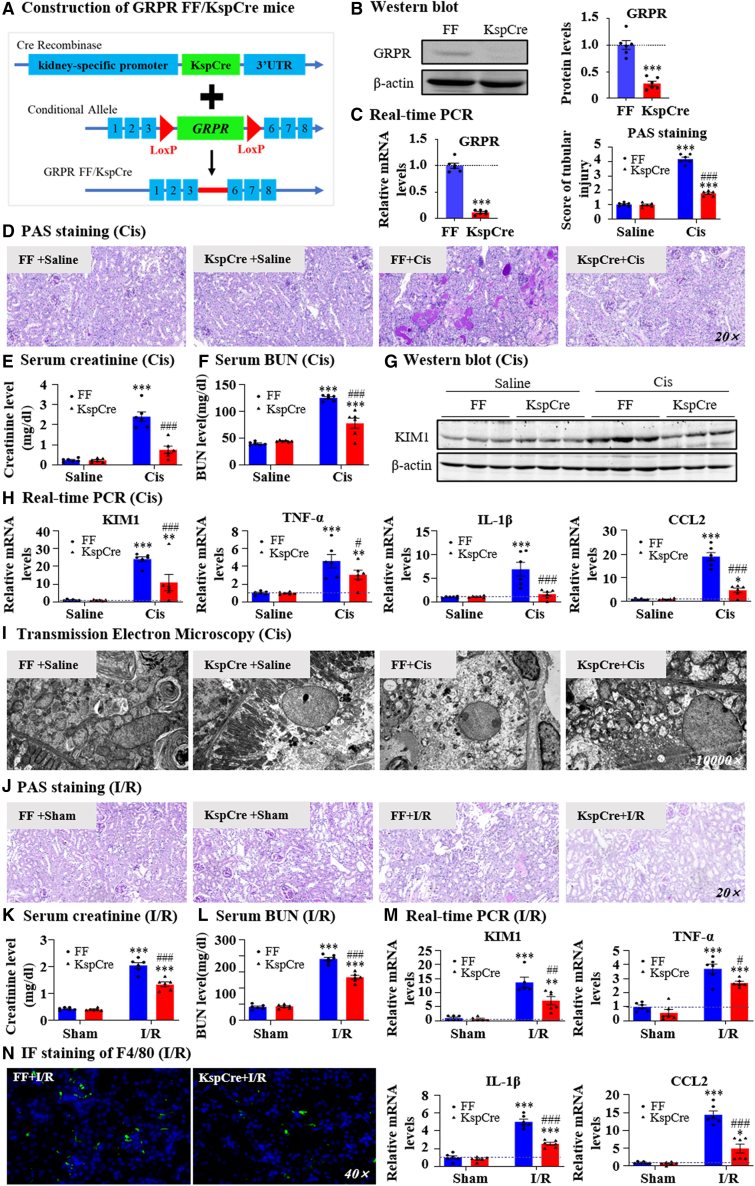
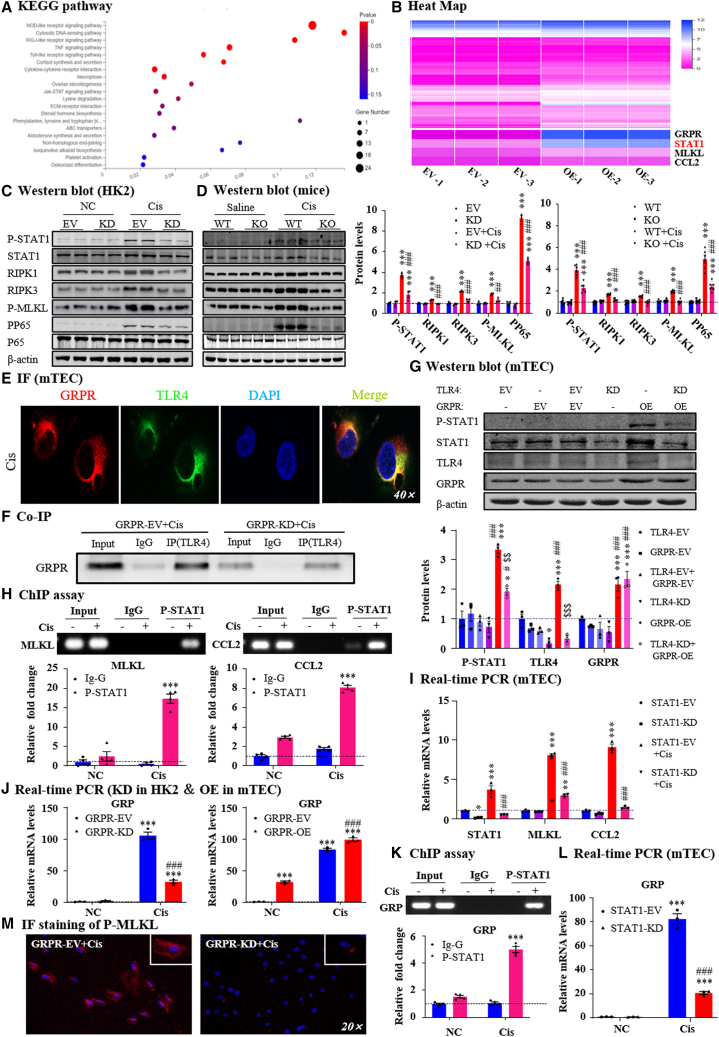
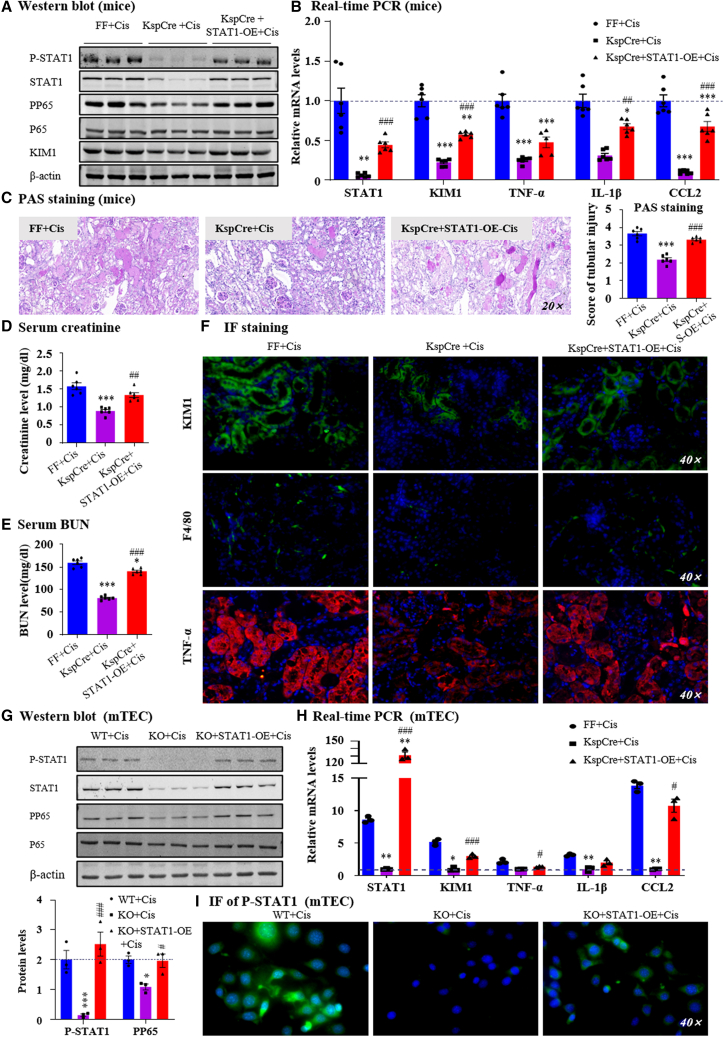
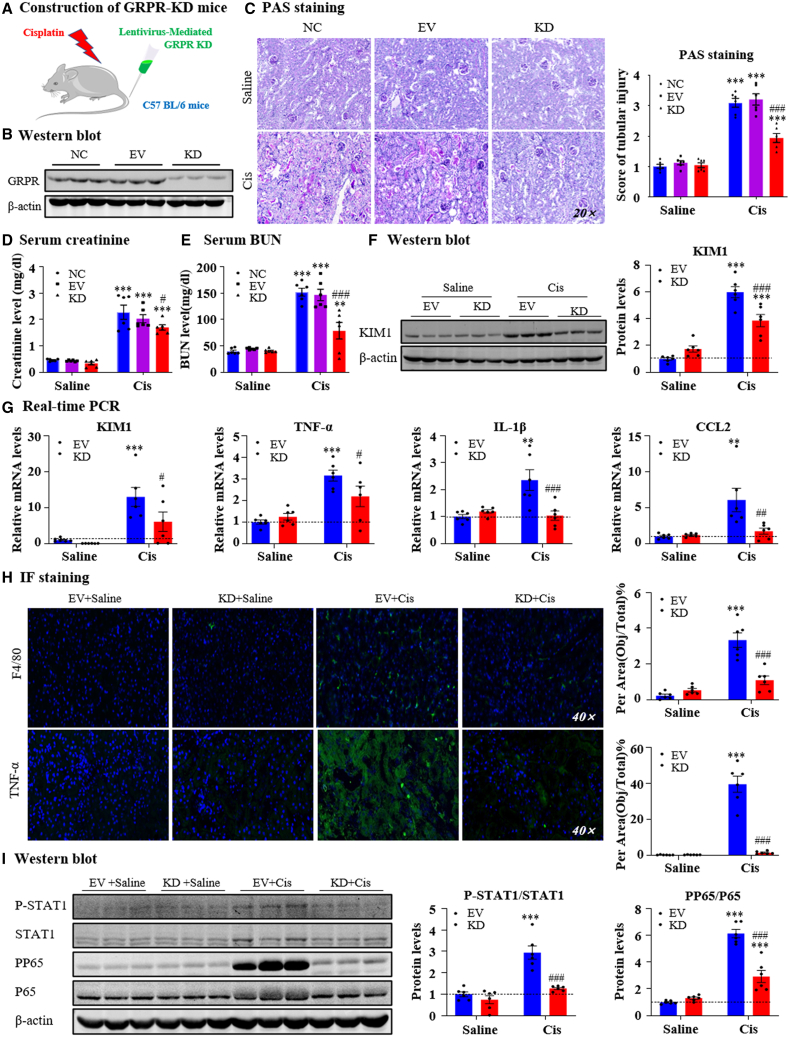
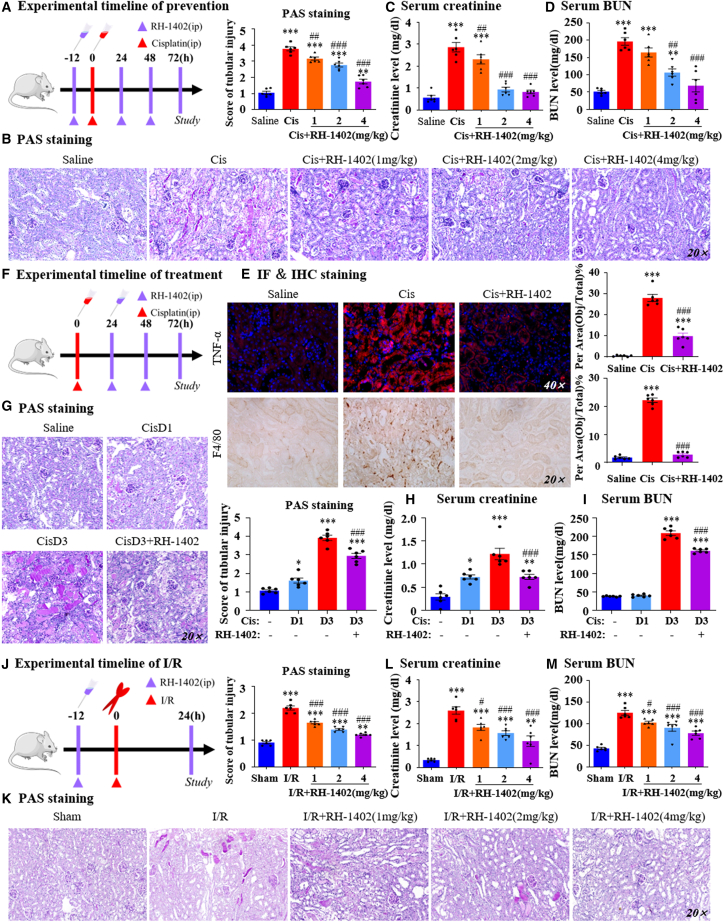
Gastrin-releasing peptide (GRP) binds to its receptor (GRP receptor [GRPR]) to regulate multiple biological processes, but the function of GRP/GRPR axis in acute kidney injury (AKI) remains unknown. In the present study, GRPR is highly expressed by tubular epithelial cells (TECs) in patients or mice with AKI, while histone deacetylase 8 may lead to the transcriptional activation of GRPR. Functionally, we uncovered that GRPR was pathogenic in AKI, as genetic deletion of GRPR was able to protect mice from cisplatin- and ischemia-induced AKI. This was further confirmed by specifically deleting the GRPR gene from TECs in GRPRFlox/Flox//KspCre mice. Mechanistically, we uncovered that GRPR was able to interact with Toll-like receptor 4 to activate STAT1 that bound the promoter of MLKL and CCL2 to induce TEC necroptosis, necroinflammation, and macrophages recruitment. This was further confirmed by overexpressing STAT1 to restore renal injury in GRPRFlox/Flox/KspCre mice. Concurrently, STAT1 induced GRP synthesis to enforce the GRP/GRPR/STAT1 positive feedback loop. Importantly, targeting GRPR by lentivirus-packaged small hairpin RNA or by treatment with a novel GRPR antagonist RH-1402 was able to inhibit cisplatin-induced AKI. In conclusion, GRPR is pathogenic in AKI and mediates AKI via the STAT1-dependent mechanism. Thus, targeting GRPR may be a novel therapeutic strategy for AKI.
Keywords: GRP; GRPR; STAT1; acute kidney injury; inflammation; necroptosis.
Copyright © 2023 The American Society of Gene and Cell Therapy. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare that they have no competing interests.
Figures

References
-
- Gao L., Zhong X., Jin J., Li J., Meng X.M. Potential targeted therapy and diagnosis based on novel insight into growth factors, receptors, and downstream effectors in acute kidney injury and acute kidney injury-chronic kidney disease progression. Signal Transduct. Target. Ther. 2020;5:9. doi: 10.1038/s41392-020-0106-1. - DOI - PMC - PubMed
-
- Wang J.N., Yang Q., Yang C., Cai Y.T., Xing T., Gao L., Wang F., Chen X., Liu X.Q., He X.Y., et al. Smad3 promotes AKI sensitivity in diabetic mice via interaction with p53 and induction of NOX4-dependent ROS production. Redox Biol. 2020;32:101479. doi: 10.1016/j.redox.2020.101479. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
