

A Systematic Analysis of the Clinical Outcome Associated with Multiple Reclassified Desmosomal Gene Variants in Arrhythmogenic Right Ventricular Cardiomyopathy Patients
- PMID: 37418234
- PMCID: PMC10721666
- DOI: 10.1007/s12265-023-10403-8
A Systematic Analysis of the Clinical Outcome Associated with Multiple Reclassified Desmosomal Gene Variants in Arrhythmogenic Right Ventricular Cardiomyopathy Patients
Abstract
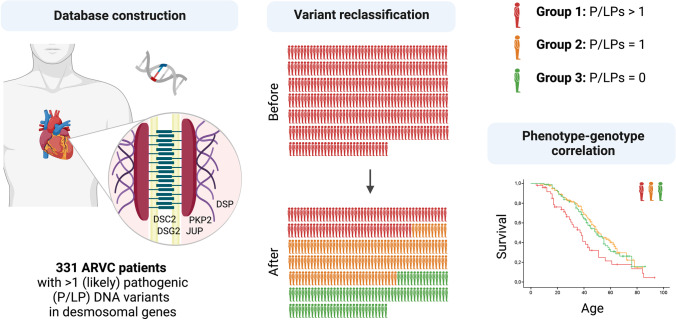
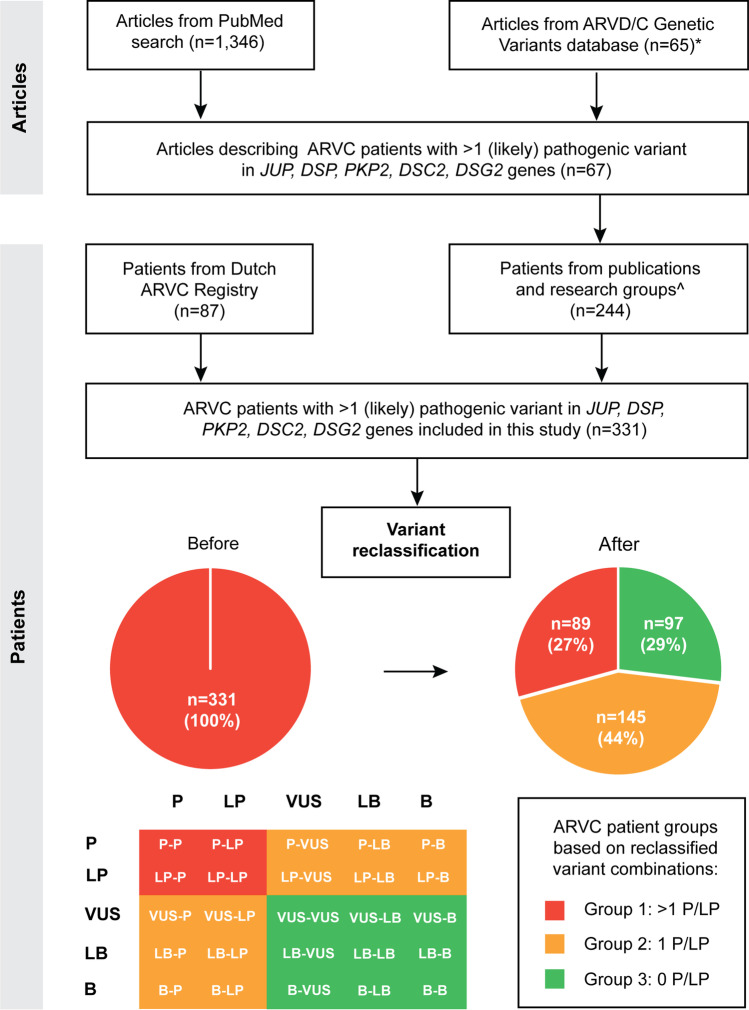
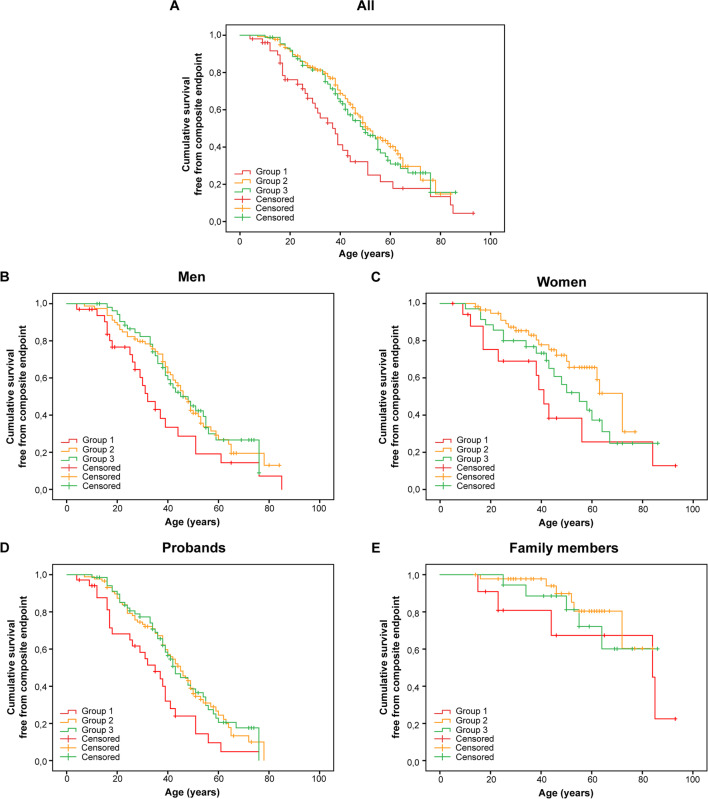
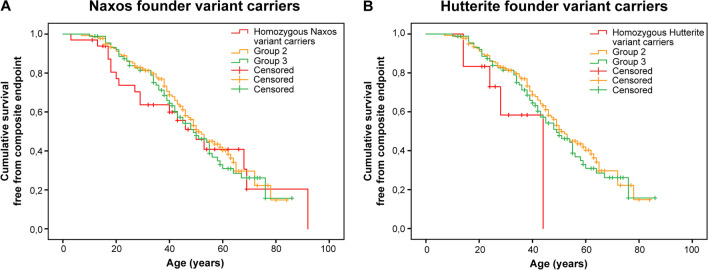
The presence of multiple pathogenic variants in desmosomal genes (DSC2, DSG2, DSP, JUP, and PKP2) in patients with arrhythmogenic right ventricular cardiomyopathy (ARVC) has been linked to a severe phenotype. However, the pathogenicity of variants is reclassified frequently, which may result in a changed clinical risk prediction. Here, we present the collection, reclassification, and clinical outcome correlation for the largest series of ARVC patients carrying multiple desmosomal pathogenic variants to date (n = 331). After reclassification, only 29% of patients remained carriers of two (likely) pathogenic variants. They reached the composite endpoint (ventricular arrhythmias, heart failure, and death) significantly earlier than patients with one or no remaining reclassified variant (hazard ratios of 1.9 and 1.8, respectively). Periodic reclassification of variants contributes to more accurate risk stratification and subsequent clinical management strategy. Graphical Abstract.
Keywords: ARVC; Arrhythmia; Composite endpoint; Desmosomal genes; Genetics; Multiple variants.
© 2023. The Author(s).
Conflict of interest statement
AMS received educational grants through his institution from Abbott, Bayer Healthcare, Biosense Webster, Biotronik, Boston Scientific, BMS/Pfizer, and Medtronic, and speaker fees from Bayer Healthcare, Daiichi-Sankyo, and Novartis. DPJ reports payments as a consultant from 4D Molecular Therapeutics, ADRx, Inc., Cytokinetics, Pfizer, and Tenaya Therapeutics outside of the scope of this work. CAJ receives salary support on a grant from Boston Scientific Corp through her institution and payment as a consultant from Pfizer and StrideBio, Inc. Consultant LQT Therapeutics (AW).
Figures
References
-
- van Tintelen JP, Van Gelder IC, Asimaki A, Suurmeijer AJ, Wiesfeld AC, Jongbloed JD, et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009;6(11):1574–1583. doi: 10.1016/j.hrthm.2009.07.041. - DOI - PubMed
-
- Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82(4):809–821. doi: 10.1016/j.ajhg.2008.01.010. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
