

Multiplex enzymatic synthesis of DNA with single-base resolution
- PMID: 37418522
- PMCID: PMC10328407
- DOI: 10.1126/sciadv.adi0263
Multiplex enzymatic synthesis of DNA with single-base resolution
Abstract
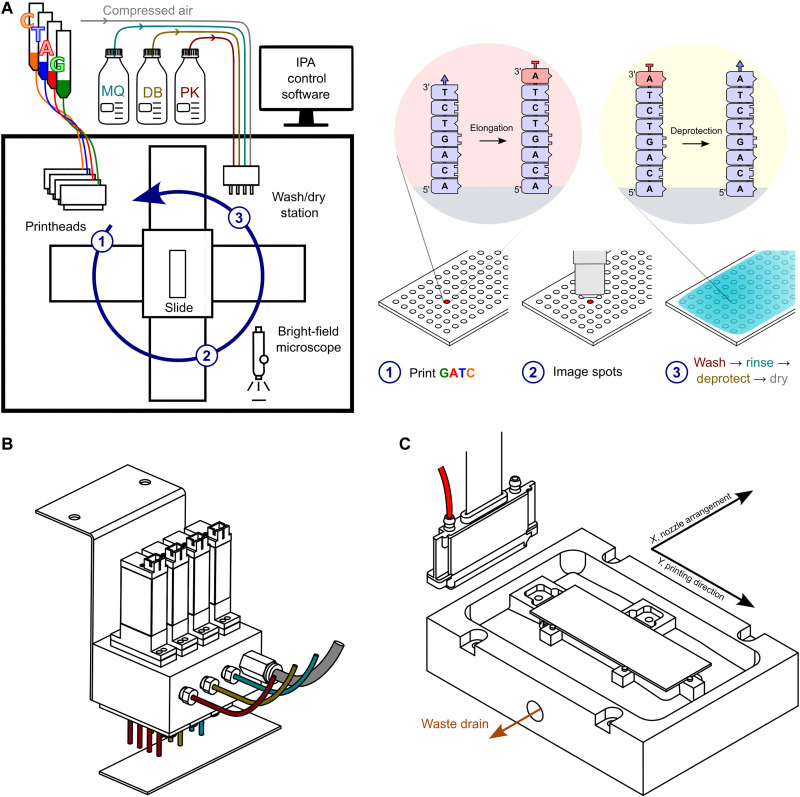
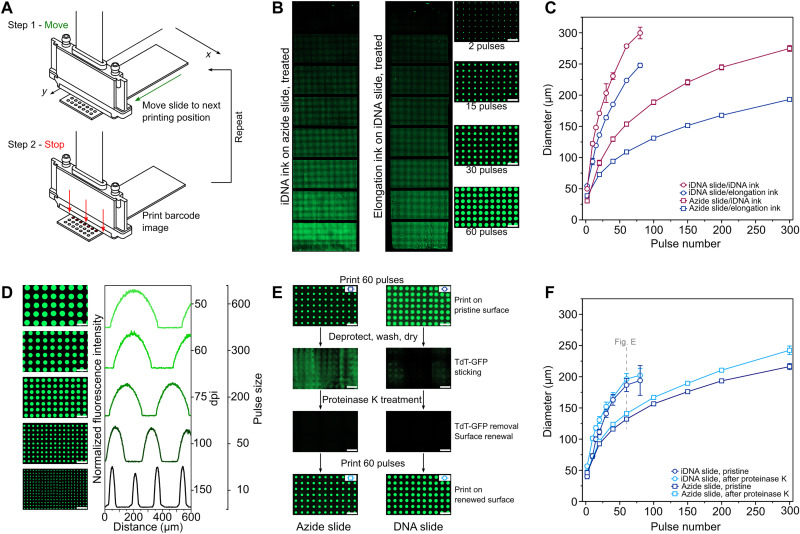
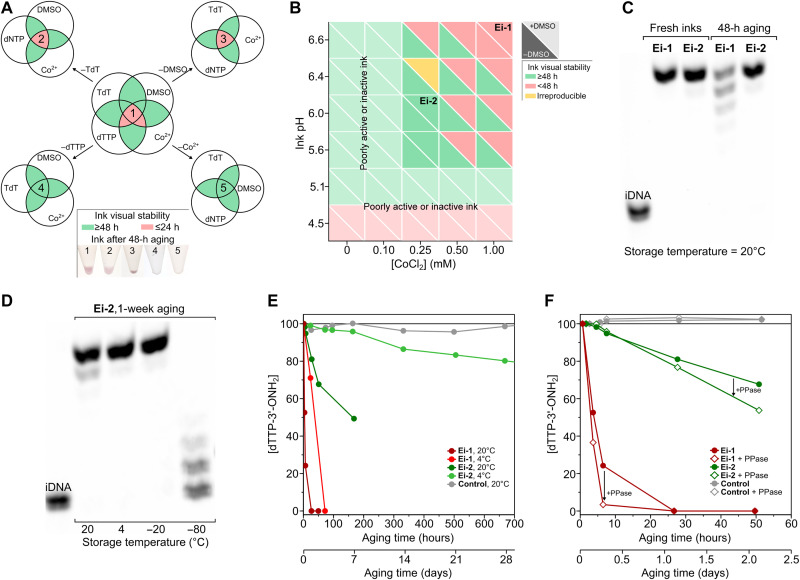
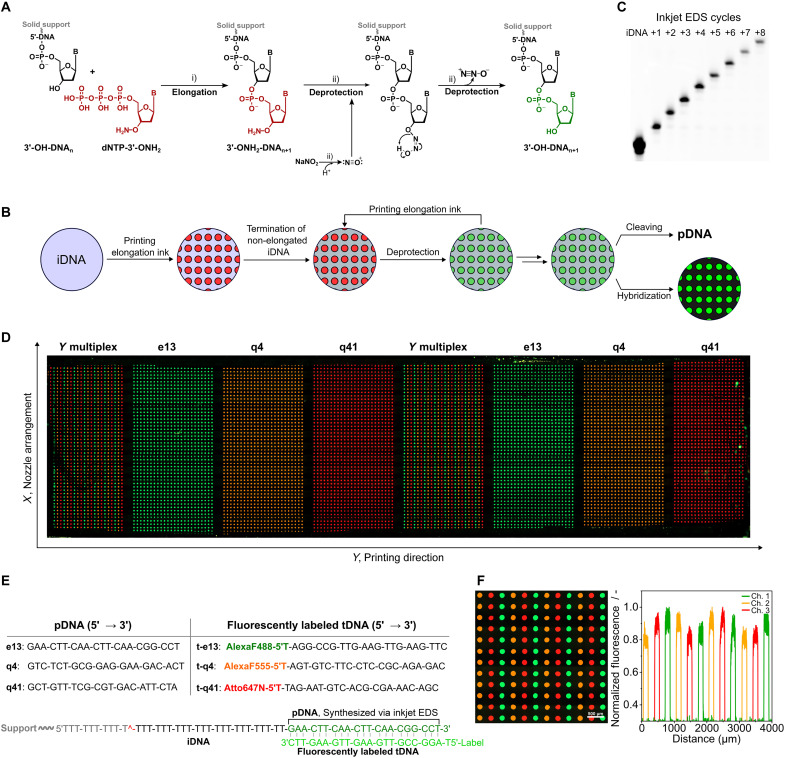
Enzymatic DNA synthesis (EDS) is a promising benchtop and user-friendly method of nucleic acid synthesis that, instead of solvents and phosphoramidites, uses mild aqueous conditions and enzymes. For applications such as protein engineering and spatial transcriptomics that require either oligo pools or arrays with high sequence diversity, the EDS method needs to be adapted and certain steps in the synthesis process spatially decoupled. Here, we have used a synthesis cycle comprising a first step of site-specific silicon microelectromechanical system inkjet dispensing of terminal deoxynucleotidyl transferase enzyme and 3' blocked nucleotide, and a second step of bulk slide washing to remove the 3' blocking group. By repeating the cycle on a substrate with an immobilized DNA primer, we show that microscale spatial control of nucleic acid sequence and length is possible, which, here, are assayed by hybridization and gel electrophoresis. This work is distinctive for enzymatically synthesizing DNA in a highly parallel manner with single base control.
Figures
Similar articles
-
Terminal deoxynucleotidyl transferase: the story of a misguided DNA polymerase.Biochim Biophys Acta. 2010 May;1804(5):1151-66. doi: 10.1016/j.bbapap.2009.06.030. Epub 2009 Jul 29. Biochim Biophys Acta. 2010. PMID: 19596089 Free PMC article. Review.
-
Evolving a terminal deoxynucleotidyl transferase for commercial enzymatic DNA synthesis.Nucleic Acids Res. 2025 Feb 8;53(4):gkaf115. doi: 10.1093/nar/gkaf115. Nucleic Acids Res. 2025. PMID: 39988321 Free PMC article.
-
Rational design of terminal deoxynucleotidyl transferase for RNA primer elongation.Int J Biol Macromol. 2025 May;309(Pt 1):142712. doi: 10.1016/j.ijbiomac.2025.142712. Epub 2025 Mar 31. Int J Biol Macromol. 2025. PMID: 40174852
-
Enhancing Terminal Deoxynucleotidyl Transferase Activity on Substrates with 3' Terminal Structures for Enzymatic De Novo DNA Synthesis.Genes (Basel). 2020 Jan 16;11(1):102. doi: 10.3390/genes11010102. Genes (Basel). 2020. PMID: 31963235 Free PMC article.
-
Applications of Terminal Deoxynucleotidyl Transferase Enzyme in Biotechnology.Chembiochem. 2023 Mar 1;24(5):e202200510. doi: 10.1002/cbic.202200510. Epub 2022 Nov 24. Chembiochem. 2023. PMID: 36342345 Review.
Cited by
-
Enzymatic synthesis of RNA oligonucleotides.Nat Biotechnol. 2025 May;43(5):691-693. doi: 10.1038/s41587-024-02322-z. Nat Biotechnol. 2025. PMID: 38997580 No abstract available.
-
Template-dependent DNA ligation for the synthesis of modified oligonucleotides.Nat Commun. 2024 Sep 13;15(1):8009. doi: 10.1038/s41467-024-52141-8. Nat Commun. 2024. PMID: 39271668 Free PMC article.
-
Controlled enzymatic synthesis of oligonucleotides.Commun Chem. 2024 Jun 18;7(1):138. doi: 10.1038/s42004-024-01216-0. Commun Chem. 2024. PMID: 38890393 Free PMC article. Review.
-
Scalable acoustic virtual stirrer for enhanced interfacial enzymatic nucleic acid reactions.Sci Adv. 2025 Mar 7;11(10):eadt6955. doi: 10.1126/sciadv.adt6955. Epub 2025 Mar 5. Sci Adv. 2025. PMID: 40043123 Free PMC article.
-
Accelerated, high-quality photolithographic synthesis of RNA microarrays in situ.Sci Adv. 2024 Aug 2;10(31):eado6762. doi: 10.1126/sciadv.ado6762. Epub 2024 Jul 31. Sci Adv. 2024. PMID: 39083603 Free PMC article.
References
-
- K. Hoff, M. Halpain, G. Garbagnati, J. S. Edwards, W. Zhou, Enzymatic synthesis of designer DNA using cyclic reversible termination and a universal template. ACS Synth. Biol. 9, 283–293 (2020). - PubMed
-
- L. Beaucage, M. H. Caruthers, Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 22, 1859–1862 (1981).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
