

Characteristics of Patients With Late-Onset Pompe Disease in France: Insights From the French Pompe Registry in 2022
- PMID: 37419682
- PMCID: PMC10501092
- DOI: 10.1212/WNL.0000000000207547
Characteristics of Patients With Late-Onset Pompe Disease in France: Insights From the French Pompe Registry in 2022
Erratum in
-
Characteristics of Patients With Late-Onset Pompe Disease in France: Insights From the French Pompe Registry in 2022.Neurology. 2024 Aug 13;103(3):e209578. doi: 10.1212/WNL.0000000000209578. Epub 2024 Jul 2. Neurology. 2024. PMID: 38954784 Free PMC article. No abstract available.
Abstract
Background and objectives: The French Pompe disease registry was created in 2004 for study of the natural course of the disease in patients. It rapidly became a major tool for assessing the long-term efficacy of enzyme replacement therapy (ERT) after the market release of alglucosidase-alfa.
Methods: Approximately 10 years after publication of the baseline characteristics of the 126 initial patients of the French Late-Onset Pompe Disease registry, we provide here an update of the clinical and biological features of patients included in this registry.
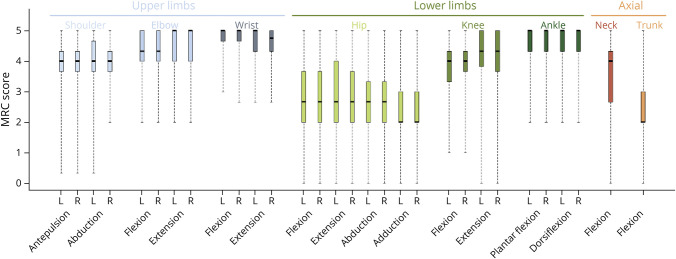
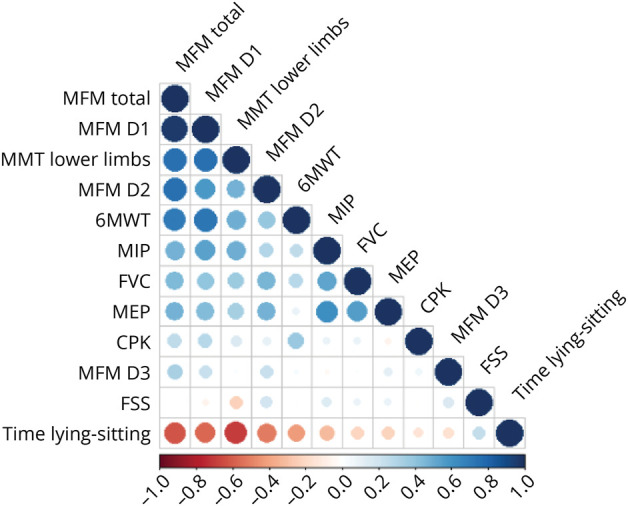
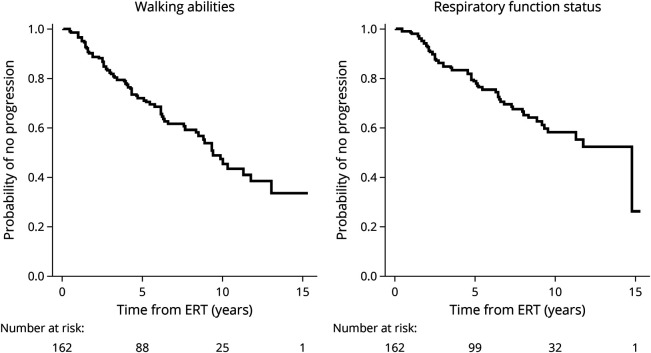
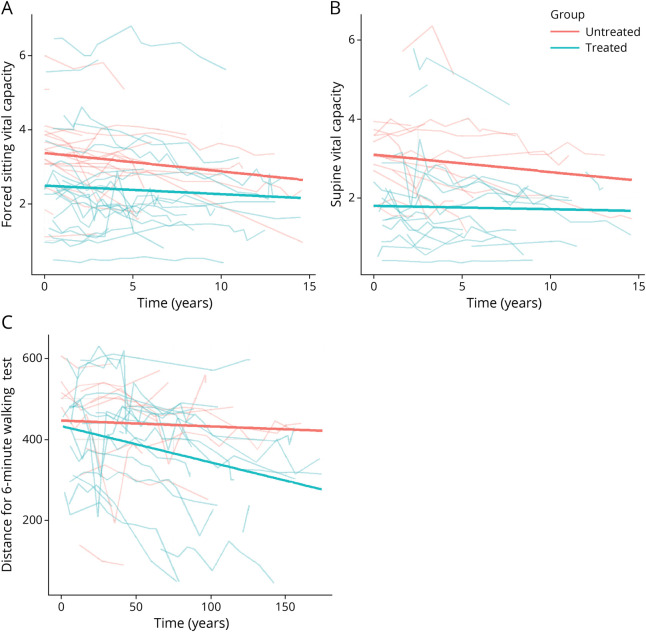
Results: We describe 210 patients followed at 31 hospital-based French neuromuscular or metabolic centers. The median age at inclusion was 48.67 ± 14.91 years. The first symptom was progressive lower limb muscle weakness, either isolated (50%) or associated with respiratory symptoms (18%), at a median age of 38 ± 14.9 years. At inclusion, 64% of the patients were able to walk independently and 14% needed a wheelchair. Positive associations were found between motor function measure, manual motor test, and 6-minute walk test (6MWT) results, and these parameters were inversely associated with the time taken to sit up from a lying position at inclusion. Seventy-two patients had been followed for at least 10 years in the registry. Thirty-three patients remained untreated a median of 12 years after symptom onset. The standard ERT dose was administered for 177 patients.
Discussion: This update confirms previous findings for the adult population included in the French Pompe disease registry, but with a lower clinical severity at inclusion, suggesting that this rare disease is now diagnosed earlier; thanks to greater awareness among physicians. The 6MWT remains an important method for assessing motor performance and walking ability. The French Pompe disease registry provides an exhaustive, nationwide overview of Pompe disease and can be used to assess individual and global responses to future treatments.
© 2023 American Academy of Neurology.
Conflict of interest statement
C. Lefeuvre received fees for consulting and sponsoring for scientific congress by Sanofi-Genzyme. F. Bouhour and E. Salort-Campana received fees for participation to scientific boards and sponsoring for scientific congress by Sanofi-Genzyme and Amicus Therapeutics. A. Behin, S. Attarian received fees for participation to scientific boards and sponsoring for scientific congress by Sanofi-Genzyme. A. Nadaj-Pakleza received fees for participation to scientific boards by Sanofi-Genzyme and Amicus Therapeutics. M. Spinazzi received fees for participation to scientific boards by Sanofi-Genzyme. C. Tard, M. Michaud, and A.-L. Bedat-Millet received sponsoring for scientific congress by Sanofi-Genzyme. J.-P. Noury received fees for participation to scientific boards by Amicus Therapeutics and sponsoring for scientific congress by Sanofi-Genzyme. P. Laforêt receive fees for participation to scientific boards by AMICUS Therapeutics, Sanofi Genzyme, Spark Therapeutics, consulting fees by Sanofi Genzyme, BioMarin, Sanofi Genzyme, Spark Therapeutics and sponsoring for scientific congress by Sanofi Genzyme, Amicus Therapeutics, Spark Therapeutics. The other authors report no relevant disclosures. Go to
Figures
References
-
- Engel AG. Acid maltase deficiency in adults: studies in four cases of a syndrome which may mimic muscular dystrophy or other myopathies. Brain. 1970;93(3):599-616. - PubMed
-
- Laforêt P, Nicolino M, Eymard PB, et al. . Juvenile and adult-onset acid maltase deficiency in France: genotype-phenotype correlation. Neurology. 2000;55(8):1122-1128. - PubMed
-
- van der Ploeg AT, Reuser AJJ. Pompe's disease. Lancet. 2008;372(9646):1342-1353. - PubMed
-
- Smith WE, Sullivan-Saarela JA, Li JS, et al. . Sibling phenotype concordance in classical infantile Pompe disease. Am J Med Genet A. 2007;143A(21):2493-2501. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical