

Atypical splicing variants in PKD1 explain most undiagnosed typical familial ADPKD
- PMID: 37419908
- PMCID: PMC10328916
- DOI: 10.1038/s41525-023-00362-z
Atypical splicing variants in PKD1 explain most undiagnosed typical familial ADPKD
Abstract
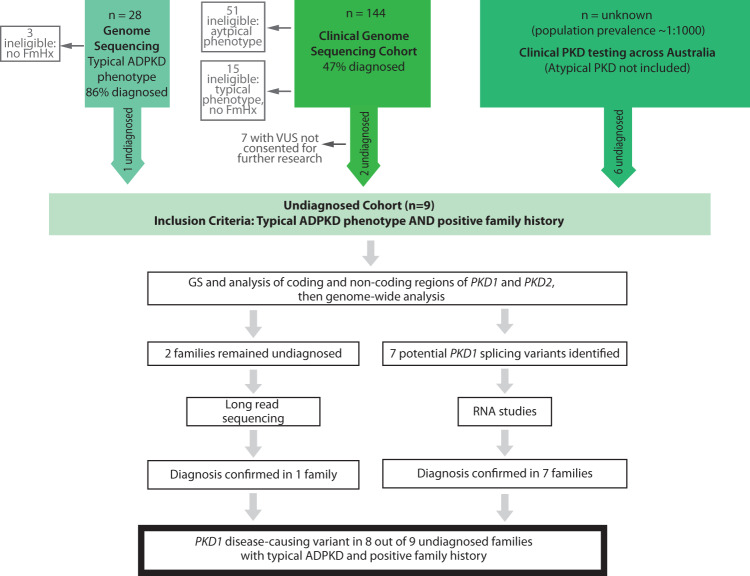
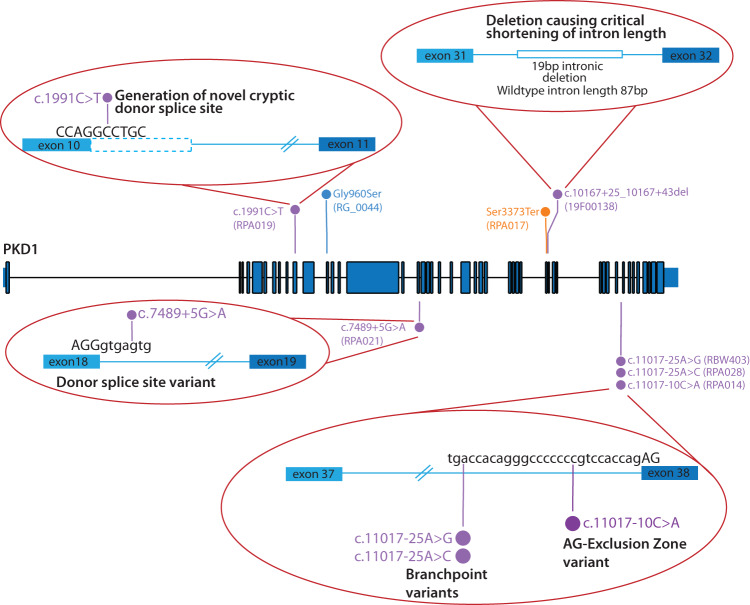
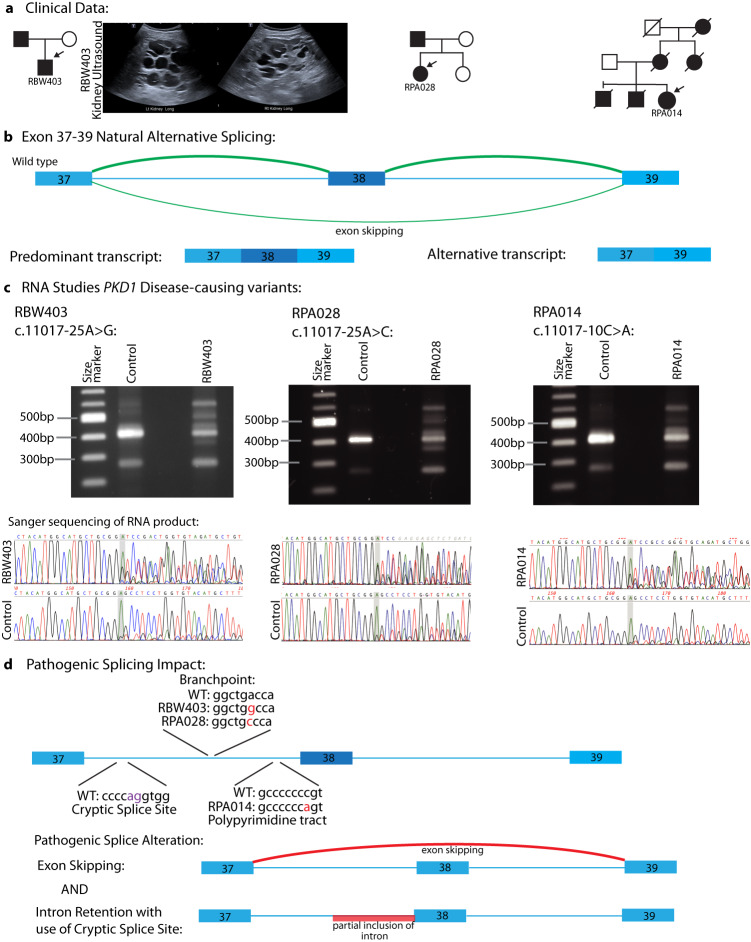
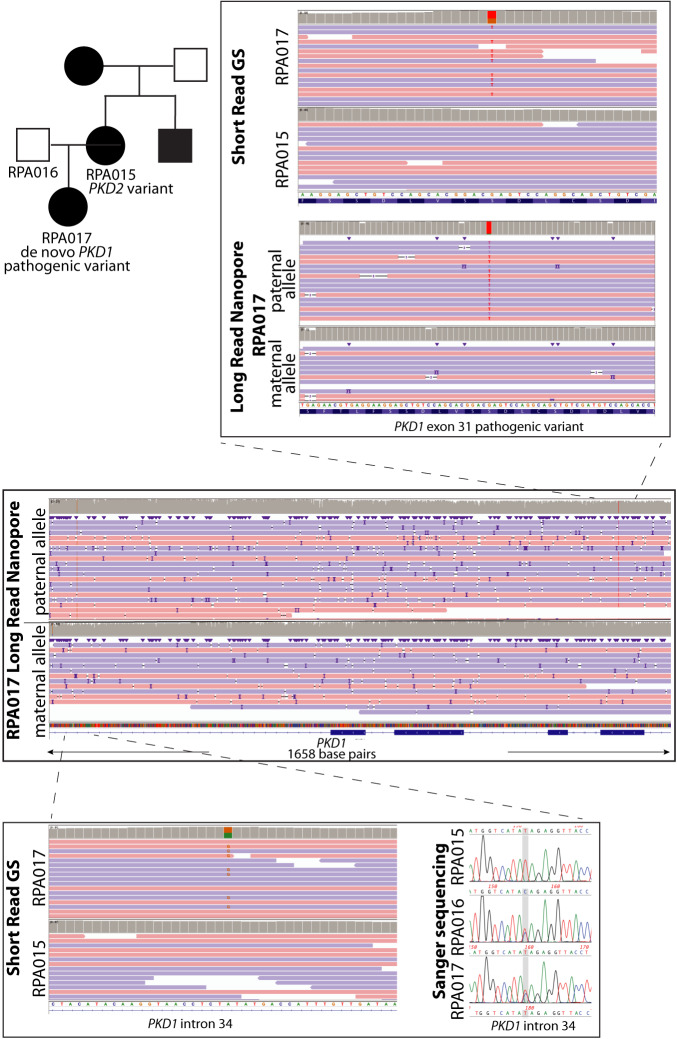
Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic cause of kidney failure and is primarily associated with PKD1 or PKD2. Approximately 10% of patients remain undiagnosed after standard genetic testing. We aimed to utilise short and long-read genome sequencing and RNA studies to investigate undiagnosed families. Patients with typical ADPKD phenotype and undiagnosed after genetic diagnostics were recruited. Probands underwent short-read genome sequencing, PKD1 and PKD2 coding and non-coding analyses and then genome-wide analysis. Targeted RNA studies investigated variants suspected to impact splicing. Those undiagnosed then underwent Oxford Nanopore Technologies long-read genome sequencing. From over 172 probands, 9 met inclusion criteria and consented. A genetic diagnosis was made in 8 of 9 (89%) families undiagnosed on prior genetic testing. Six had variants impacting splicing, five in non-coding regions of PKD1. Short-read genome sequencing identified novel branchpoint, AG-exclusion zone and missense variants generating cryptic splice sites and a deletion causing critical intron shortening. Long-read sequencing confirmed the diagnosis in one family. Most undiagnosed families with typical ADPKD have splice-impacting variants in PKD1. We describe a pragmatic method for diagnostic laboratories to assess PKD1 and PKD2 non-coding regions and validate suspected splicing variants through targeted RNA studies.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
Miscellaneous
