

The use of RNA-based treatments in the field of cancer immunotherapy
- PMID: 37420174
- PMCID: PMC10401791
- DOI: 10.1186/s12943-023-01807-w
The use of RNA-based treatments in the field of cancer immunotherapy
Abstract
Over the past several decades, mRNA vaccines have evolved from a theoretical concept to a clinical reality. These vaccines offer several advantages over traditional vaccine techniques, including their high potency, rapid development, low-cost manufacturing, and safe administration. However, until recently, concerns over the instability and inefficient distribution of mRNA in vivo have limited their utility. Fortunately, recent technological advancements have mostly resolved these concerns, resulting in the development of numerous mRNA vaccination platforms for infectious diseases and various types of cancer. These platforms have shown promising outcomes in both animal models and humans. This study highlights the potential of mRNA vaccines as a promising alternative approach to conventional vaccine techniques and cancer treatment. This review article aims to provide a thorough and detailed examination of mRNA vaccines, including their mechanisms of action and potential applications in cancer immunotherapy. Additionally, the article will analyze the current state of mRNA vaccine technology and highlight future directions for the development and implementation of this promising vaccine platform as a mainstream therapeutic option. The review will also discuss potential challenges and limitations of mRNA vaccines, such as their stability and in vivo distribution, and suggest ways to overcome these issues. By providing a comprehensive overview and critical analysis of mRNA vaccines, this review aims to contribute to the advancement of this innovative approach to cancer treatment.
Keywords: Animal models; Cancer immunotherapy; Clinical trials; Infectious diseases; Messenger RNA; Technological advancements; Therapeutic use; mRNA vaccines.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
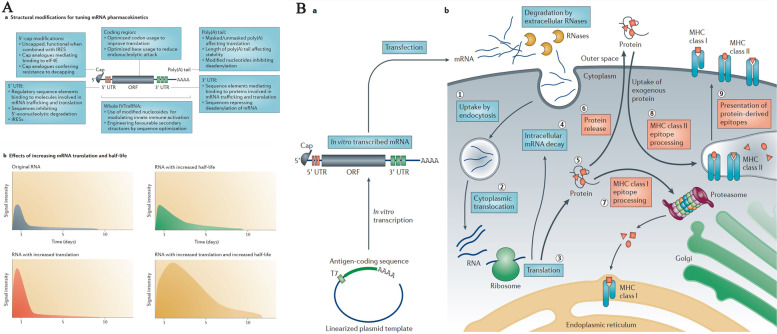
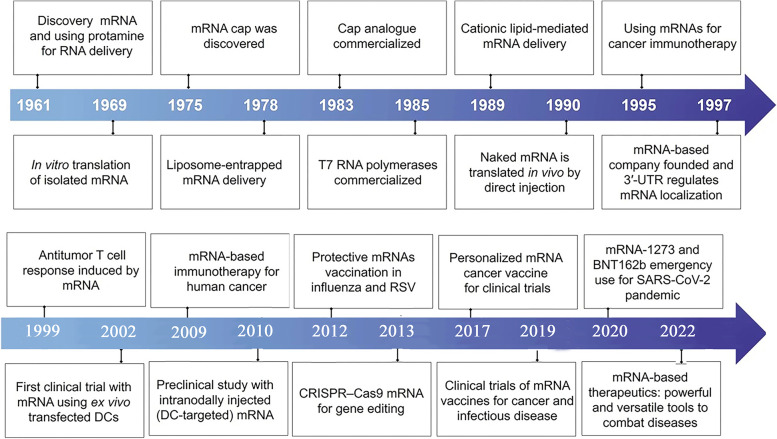
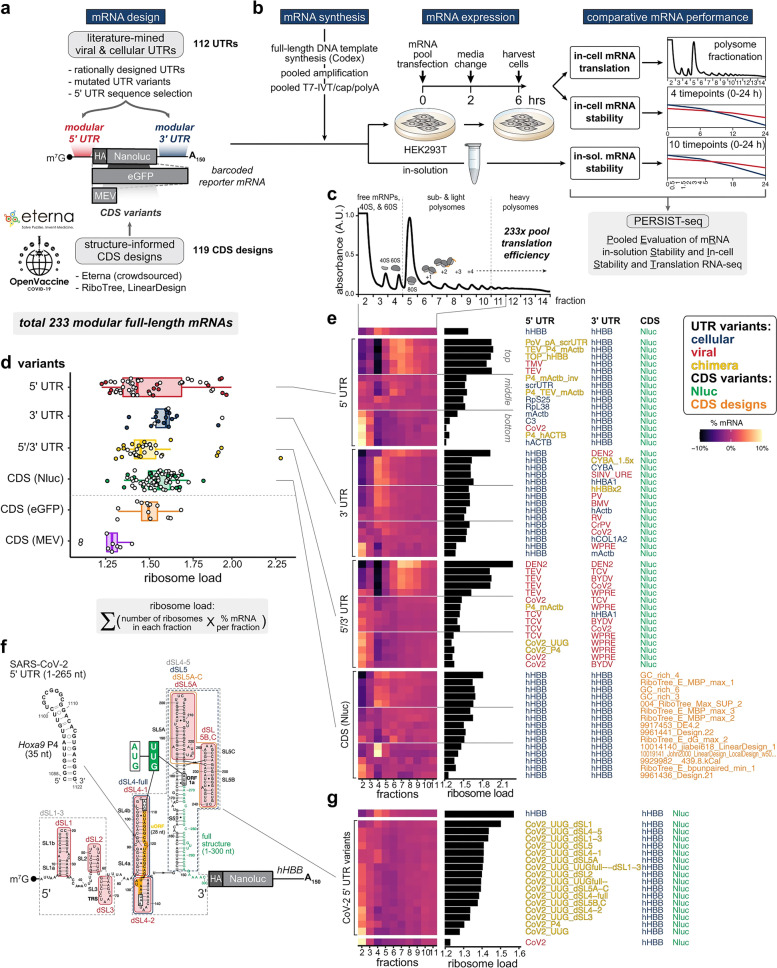
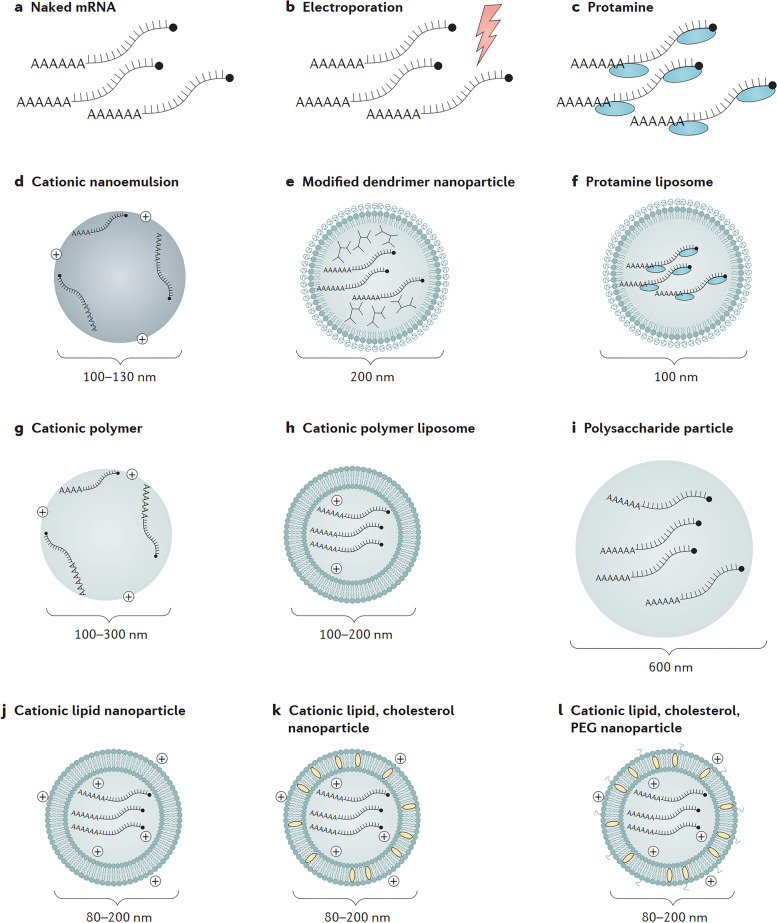
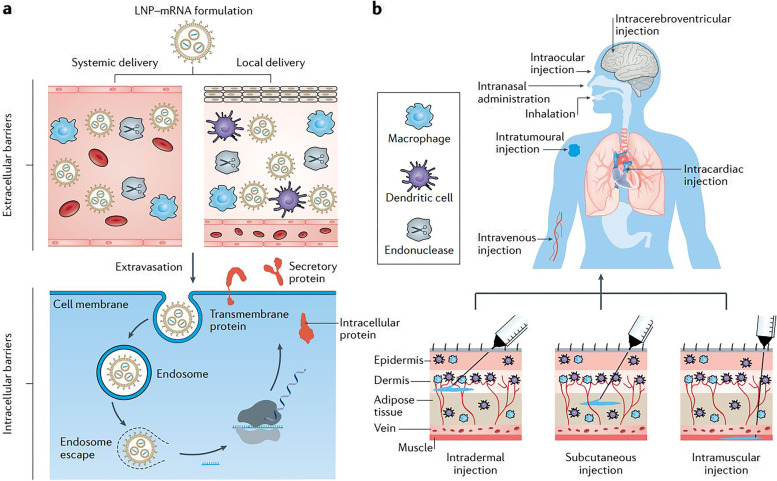
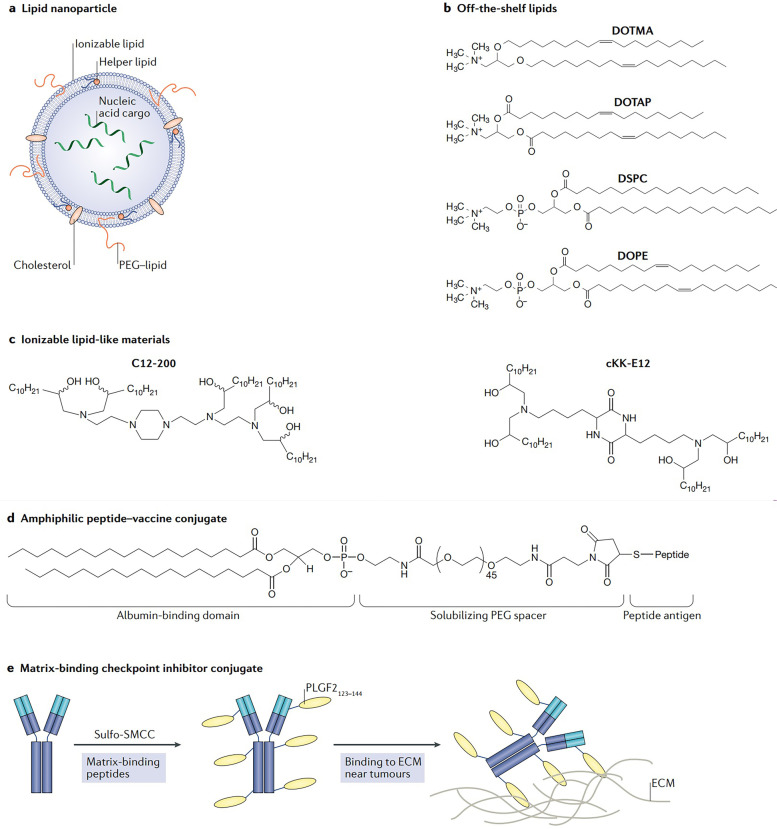
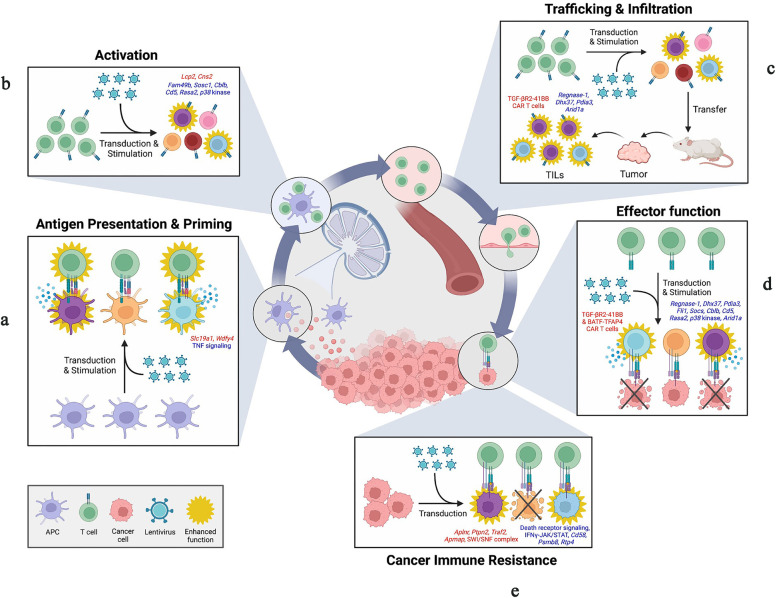
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
