

Integrative meta-omics in Galaxy and beyond
- PMID: 37420292
- PMCID: PMC10329324
- DOI: 10.1186/s40793-023-00514-9
Integrative meta-omics in Galaxy and beyond
Abstract
Background: 'Omics methods have empowered scientists to tackle the complexity of microbial communities on a scale not attainable before. Individually, omics analyses can provide great insight; while combined as "meta-omics", they enhance the understanding of which organisms occupy specific metabolic niches, how they interact, and how they utilize environmental nutrients. Here we present three integrative meta-omics workflows, developed in Galaxy, for enhanced analysis and integration of metagenomics, metatranscriptomics, and metaproteomics, combined with our newly developed web-application, ViMO (Visualizer for Meta-Omics) to analyse metabolisms in complex microbial communities.
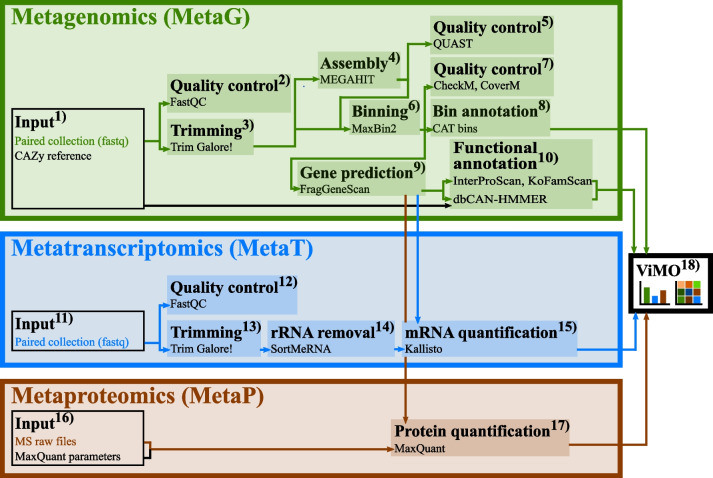
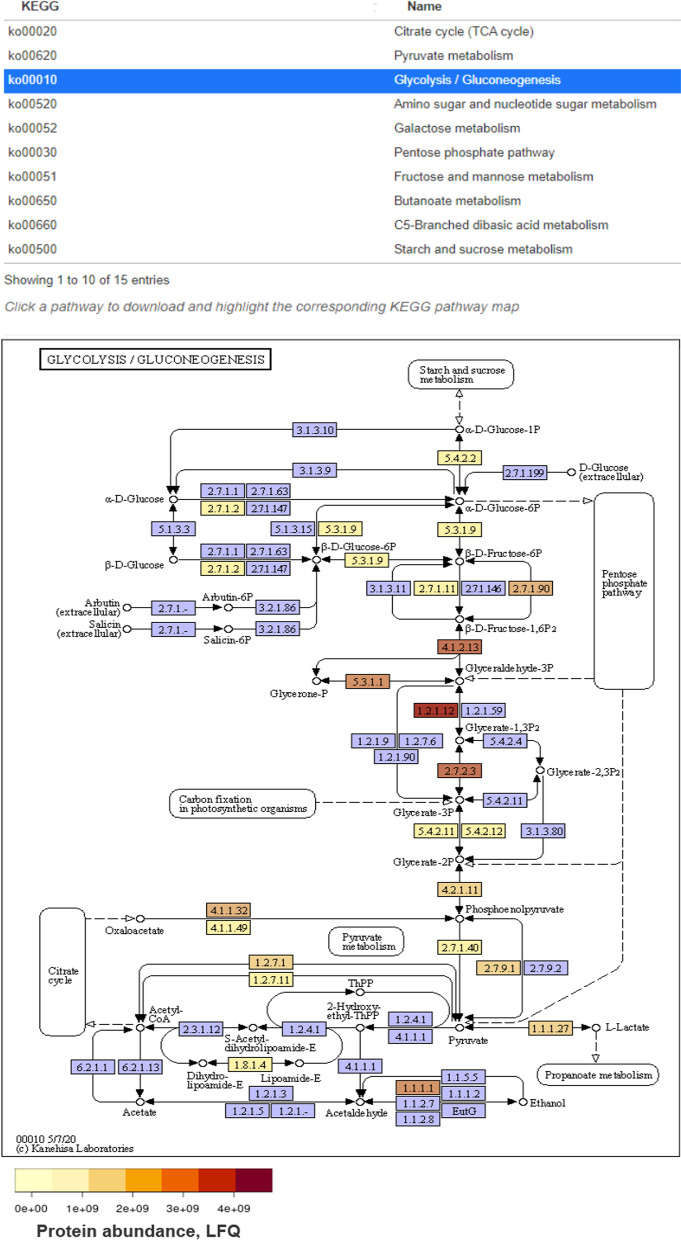
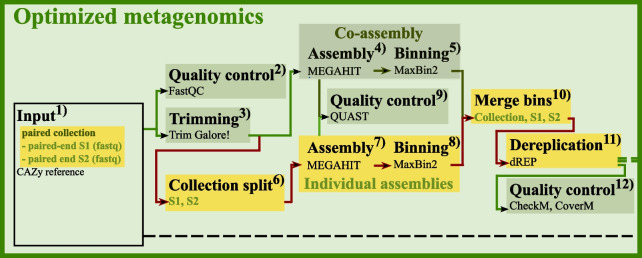
Results: In this study, we applied the workflows on a highly efficient cellulose-degrading minimal consortium enriched from a biogas reactor to analyse the key roles of uncultured microorganisms in complex biomass degradation processes. Metagenomic analysis recovered metagenome-assembled genomes (MAGs) for several constituent populations including Hungateiclostridium thermocellum, Thermoclostridium stercorarium and multiple heterogenic strains affiliated to Coprothermobacter proteolyticus. The metagenomics workflow was developed as two modules, one standard, and one optimized for improving the MAG quality in complex samples by implementing a combination of single- and co-assembly, and dereplication after binning. The exploration of the active pathways within the recovered MAGs can be visualized in ViMO, which also provides an overview of the MAG taxonomy and quality (contamination and completeness), and information about carbohydrate-active enzymes (CAZymes), as well as KEGG annotations and pathways, with counts and abundances at both mRNA and protein level. To achieve this, the metatranscriptomic reads and metaproteomic mass-spectrometry spectra are mapped onto predicted genes from the metagenome to analyse the functional potential of MAGs, as well as the actual expressed proteins and functions of the microbiome, all visualized in ViMO.
Conclusion: Our three workflows for integrative meta-omics in combination with ViMO presents a progression in the analysis of 'omics data, particularly within Galaxy, but also beyond. The optimized metagenomics workflow allows for detailed reconstruction of microbial community consisting of MAGs with high quality, and thus improves analyses of the metabolism of the microbiome, using the metatranscriptomics and metaproteomics workflows.
Keywords: Bioinformatics; Galaxy; Integrated meta-omics; Metagenomics; Metaproteomics; Metatrascriptomics.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials