

LSMMD-MA: scaling multimodal data integration for single-cell genomics data analysis
- PMID: 37421399
- PMCID: PMC10336029
- DOI: 10.1093/bioinformatics/btad420
LSMMD-MA: scaling multimodal data integration for single-cell genomics data analysis
Abstract
Motivation: Modality matching in single-cell omics data analysis-i.e. matching cells across datasets collected using different types of genomic assays-has become an important problem, because unifying perspectives across different technologies holds the promise of yielding biological and clinical discoveries. However, single-cell dataset sizes can now reach hundreds of thousands to millions of cells, which remain out of reach for most multimodal computational methods.
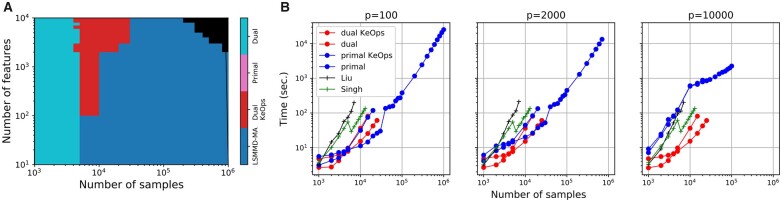
Results: We propose LSMMD-MA, a large-scale Python implementation of the MMD-MA method for multimodal data integration. In LSMMD-MA, we reformulate the MMD-MA optimization problem using linear algebra and solve it with KeOps, a CUDA framework for symbolic matrix computation in Python. We show that LSMMD-MA scales to a million cells in each modality, two orders of magnitude greater than existing implementations.
Availability and implementation: LSMMD-MA is freely available at https://github.com/google-research/large_scale_mmdma and archived at https://doi.org/10.5281/zenodo.8076311.
© The Author(s) 2023. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures
References
-
- Abadi M, Agarwal A, Barham P. et al. TensorFlow: large-scale machine learning on heterogeneous systems. [Computer software]. arXiv preprint arXiv:1603.04467, 2016. https://www.tensorflow.org.
-
- Charlier B, Feydy J, Glaunès JA. et al. Kernel operations on the GPU, with autodiff, without memory overflows. J Mach Learn Res 2021;22:1–6.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
