

DNA 5-methylcytosine detection and methylation phasing using PacBio circular consensus sequencing
- PMID: 37422489
- PMCID: PMC10329642
- DOI: 10.1038/s41467-023-39784-9
DNA 5-methylcytosine detection and methylation phasing using PacBio circular consensus sequencing
Abstract
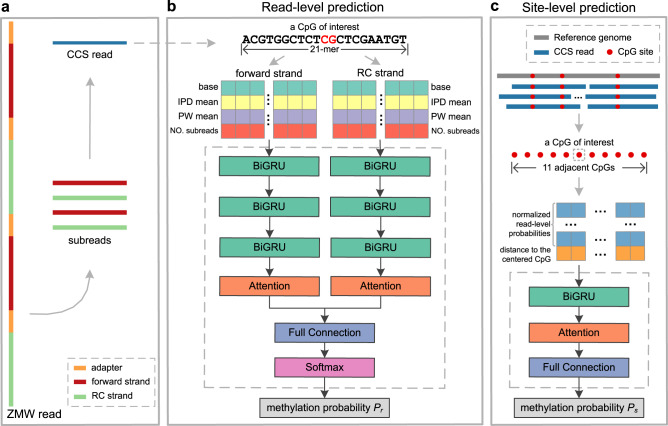
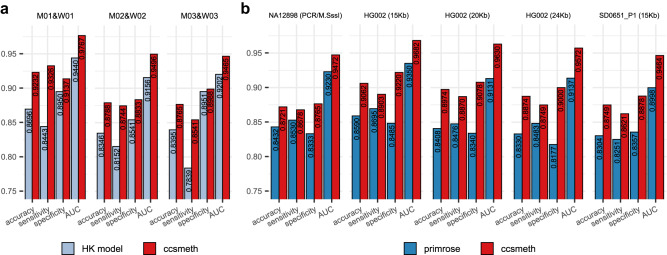
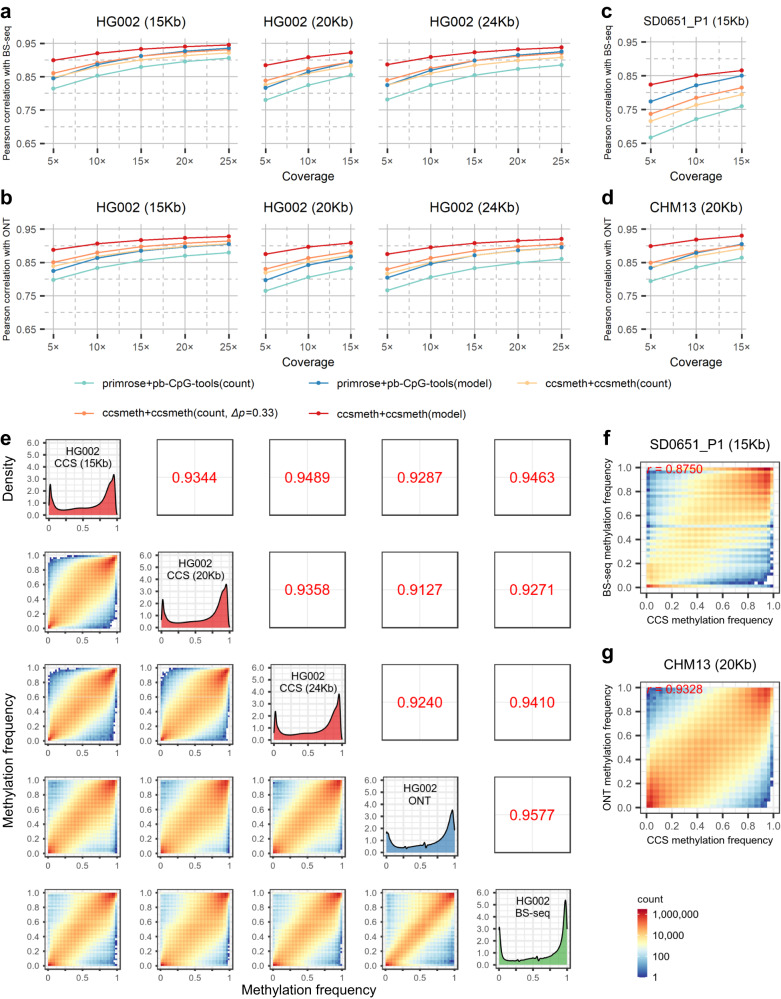
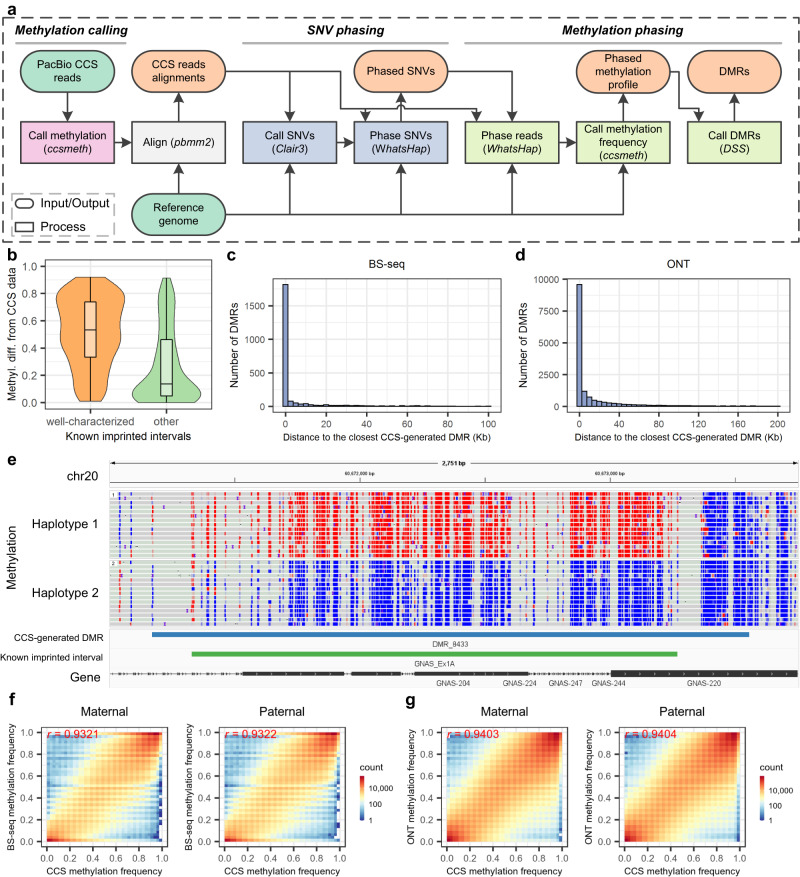
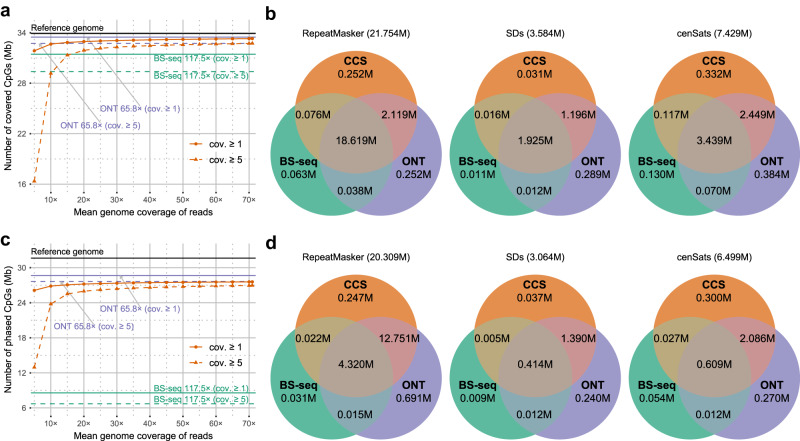
Long single-molecular sequencing technologies, such as PacBio circular consensus sequencing (CCS) and nanopore sequencing, are advantageous in detecting DNA 5-methylcytosine in CpGs (5mCpGs), especially in repetitive genomic regions. However, existing methods for detecting 5mCpGs using PacBio CCS are less accurate and robust. Here, we present ccsmeth, a deep-learning method to detect DNA 5mCpGs using CCS reads. We sequence polymerase-chain-reaction treated and M.SssI-methyltransferase treated DNA of one human sample using PacBio CCS for training ccsmeth. Using long (≥10 Kb) CCS reads, ccsmeth achieves 0.90 accuracy and 0.97 Area Under the Curve on 5mCpG detection at single-molecule resolution. At the genome-wide site level, ccsmeth achieves >0.90 correlations with bisulfite sequencing and nanopore sequencing using only 10× reads. Furthermore, we develop a Nextflow pipeline, ccsmethphase, to detect haplotype-aware methylation using CCS reads, and then sequence a Chinese family trio to validate it. ccsmeth and ccsmethphase can be robust and accurate tools for detecting DNA 5-methylcytosines.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
