

The Role of Glutamate Underlying Treatment-resistant Depression
- PMID: 37424412
- PMCID: PMC10335903
- DOI: 10.9758/cpn.22.1034
The Role of Glutamate Underlying Treatment-resistant Depression
Abstract
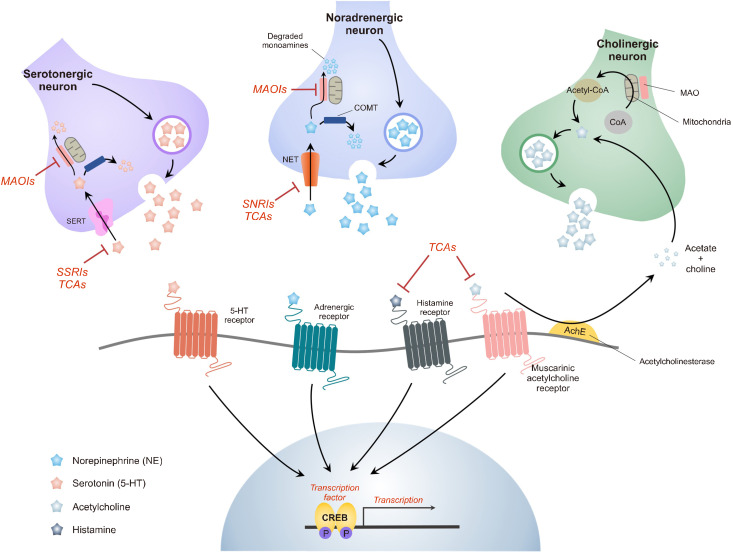
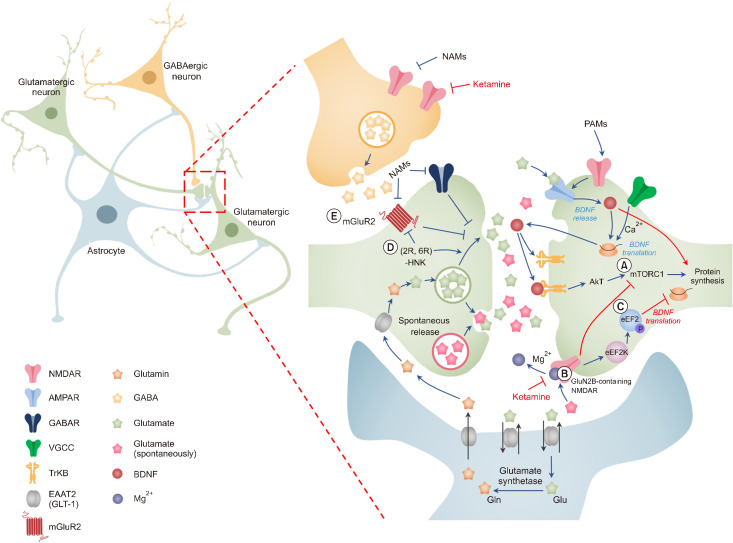
The monoamine hypothesis has significantly improved our understanding of mood disorders and their treatment by linking monoaminergic abnormalities to the pathophysiology of mood disorders. Even 50 years after the monoamine hypothesis was established, some patients do not respond to treatments for depression, including selective serotonin reuptake drugs. Accumulating evidence shows that patients with treatment-resistant depression (TRD) have severe abnormalities in the neuroplasticity and neurotrophic factor pathways, indicating that different treatment approaches may be necessary. Therefore, the glutamate hypothesis is gaining attention as a novel hypothesis that can overcome monoamine restrictions. Glutamate has been linked to structural and maladaptive morphological alterations in several brain areas associated with mood disorders. Recently, ketamine, an N-methyl-D-aspartate receptor (NMDAR) antagonist, has shown efficacy in TRD treatment and has received the U.S. Food and Drug Administration approval, revitalizing psychiatry research. However, the mechanism by which ketamine improves TRD remains unclear. In this review, we re-examined the glutamate hypothesis, bringing the glutamate system onboard to join the modulation of the monoamine systems, emphasizing the most prominent ketamine antidepressant mechanisms, such as NMDAR inhibition and NMDAR disinhibition in GABAergic interneurons. Furthermore, we discuss the animal models used in preclinical studies and the sex differences in the effects of ketamine.
Keywords: Chronic stress; Glutamate hypothesis; Ketamine; Models; Sex characteristics; Treatment-resistant depression; animal.
Conflict of interest statement
No potential conflict of interest relevant to this article was reported.
Figures

References
Publication types
LinkOut - more resources
Full Text Sources
